From Data to Dynamics: How to Integrate Omics with Flux Balance Analysis for Predictive Systems Biology
This article provides a comprehensive guide for researchers and drug development professionals on integrating multi-omics data with Flux Balance Analysis (FBA) to construct predictive, genome-scale metabolic models.
From Data to Dynamics: How to Integrate Omics with Flux Balance Analysis for Predictive Systems Biology
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on integrating multi-omics data with Flux Balance Analysis (FBA) to construct predictive, genome-scale metabolic models. It covers foundational concepts, modern methodological pipelines for constraint integration, common pitfalls and optimization strategies, and critical validation frameworks. By bridging high-throughput molecular data with computational modeling, this integration enables the prediction of metabolic phenotypes, identification of drug targets, and discovery of novel biomarkers, advancing both basic research and translational applications.
The Essential Bridge: Understanding Omics and Flux Balance Analysis Fundamentals
What is Flux Balance Analysis? A Primer on Constraint-Based Modeling.
Flux Balance Analysis (FBA) is a cornerstone mathematical approach within Constraint-Based Modeling (CBM) used to predict metabolic flux distributions in biological systems. Operating under the assumption of steady-state mass balance, FBA employs linear programming to optimize an objective function (e.g., biomass production or ATP synthesis) within the confines of a genome-scale metabolic reconstruction (GEM). Within the thesis context of "Integrating omics data with flux balance analysis research," FBA serves as the computational scaffold upon which multi-omics layers—such as transcriptomics, proteomics, and metabolomics—are integrated to generate context-specific, predictive models of cellular physiology for applications in metabolic engineering and drug target discovery.
Core Principles and Quantitative Framework
FBA is governed by key constraints derived from physicochemical laws. The fundamental equation is:
S · v = 0
Where S is the m x n stoichiometric matrix (m metabolites, n reactions), and v is the vector of reaction fluxes. This represents the steady-state constraint, ensuring internal metabolite concentrations do not change.
Additional constraints are applied: α ≤ v ≤ β where α and β are lower and upper bounds for each reaction flux, often based on known enzyme capacities (Vmax) or uptake rates.
The system then solves for v that maximizes/minimizes a linear objective function Z = cᵀ·v, where c is a vector of weights, commonly defining a biomass reaction.
Table 1: Key Constraints in a Standard FBA Problem
| Constraint Type | Mathematical Representation | Biological Interpretation |
|---|---|---|
| Steady-State | S · v = 0 | Internal metabolites are neither created nor destroyed. |
| Capacity | αi ≤ vi ≤ β_i | Enzymatic reaction rates are limited by kinetic and thermodynamic factors. |
| Objective | Maximize/Minimize cᵀ·v | Cell is optimizing for a goal (e.g., growth, product synthesis). |
Protocol: Performing a Basic Flux Balance Analysis
Objective: To predict the optimal growth rate and flux distribution of E. coli under aerobic, glucose-limited conditions using a genome-scale model.
Materials & Reagents:
- Software: COBRA Toolbox (MATLAB), cobrapy (Python), or similar.
- Model: A curated genome-scale metabolic reconstruction (e.g., E. coli iJO1366).
- Solver: A linear programming solver (e.g., GLPK, IBM CPLEX, Gurobi).
Procedure:
- Model Acquisition & Import: Download a validated model in SBML format. Load it into your chosen software environment (e.g., using
readCbModelin COBRA Toolbox). - Define Medium Constraints: Set the lower bound of the glucose exchange reaction (e.g.,
EX_glc__D_e) to -10 mmol/gDW/hr (negative denotes uptake). Set oxygen uptake (EX_o2_e) to allow free flux (e.g., -20 to 0). Set bounds for all other carbon sources to 0. - Set Objective Function: Assign the biomass reaction (e.g.,
BIOMASS_Ec_iJO1366_core_53p95M) as the objective to be maximized. - Run FBA: Execute the linear programming optimization (e.g., using
optimizeCbModel). The solver will find a flux distribution that satisfies all constraints and maximizes biomass production. - Extract Results: Capture the optimal growth rate (value of the objective function) and the flux through each reaction in the network.
- Validation: Compare the predicted growth rate and by-product secretion (e.g., acetate) against known experimental data from literature.
Integration with Omics Data: Protocol for Generating a Context-Specific Model
Objective: Integrate RNA-Seq transcriptomic data to construct a tissue-specific liver metabolic model.
Procedure:
- Data Preparation: Obtain a high-quality human generic GEM (e.g., Recon3D) and liver-specific RNA-Seq data (TPM or FPKM values).
- Gene-Protein-Reaction (GPR) Mapping: Use the Boolean GPR rules in the model to map gene identifiers to the corresponding metabolic reactions.
- Expression Thresholding: Define a threshold (e.g., median expression, percentile) to classify reactions as "active" or "inactive" based on the expression of associated genes.
- Apply Constraints: Use an algorithm like GIMME, iMAT, or FASTCORE.
- For iMAT: Apply
flux >= εfor highly expressed reactions andflux = 0for very lowly expressed reactions, whereεis a small positive flux. The objective is to maximize the number of reactions carrying flux consistent with expression status.
- For iMAT: Apply
- Model Extraction: Solve the resulting mixed-integer linear programming (MILP) or linear programming (LP) problem to obtain a functional, context-specific subnetwork.
- Gap-Filling & Validation: Use a biochemical database to add minimal missing reactions essential for network connectivity. Validate model functionality by ensuring it can produce key liver metabolites (e.g., urea, albumin precursors) under physiological conditions.
The Scientist's Toolkit
Table 2: Essential Research Reagents & Resources for FBA
| Item / Resource | Type | Function / Purpose |
|---|---|---|
| COBRA Toolbox | Software Suite | A MATLAB toolbox providing standardized functions for CBM, including FBA, model parsing, and simulation. |
| cobrapy | Software Library | A Python package for CBM, offering a flexible, scriptable alternative to COBRA Toolbox. |
| BiGG Models Database | Online Repository | A curated collection of high-quality, genome-scale metabolic reconstructions in a standardized namespace. |
| MetaNetX | Online Platform | A resource for model repository, analysis, and biochemical network reconciliation across namespaces. |
| SBML (Systems Biology Markup Language) | Format | An XML-based interchange format for computational models; essential for sharing and publishing models. |
| GLPK / CPLEX / Gurobi | Solver Software | Mathematical optimization solvers used to compute the linear programming solution at the heart of FBA. |
| MEMOTE | Software Tool | An open-source test suite for standardized and reproducible quality assessment of genome-scale metabolic models. |
Visualizations
Title: FBA and Omics Integration Workflow
Title: Simple Metabolic Network for FBA
The integration of multi-omics data with Flux Balance Analysis (FBA) enables the construction of genome-scale, context-specific metabolic models. This paradigm is critical for elucidating disease mechanisms, identifying drug targets, and advancing systems biology. The application notes below detail the role of each omics layer in this integrative framework.
Table 1: Omics Data Types and Their Contribution to Constraint-Based Metabolic Modeling
| Omics Layer | Primary Measurement | Key Technology | Use in FBA Integration | Typical Data Scale |
|---|---|---|---|---|
| Genomics | DNA Sequence & Variation | Whole Genome Sequencing, SNP Arrays | Defines gene repertoire (GPR rules) and model reconstruction. | 3.2 Gb (human genome) |
| Transcriptomics | RNA Abundance (mRNA) | RNA-Seq, Microarrays | Informs gene state (on/off) via expression thresholds (e.g., >1 TPM). Can constrain reaction bounds. | 20,000-25,000 genes |
| Proteomics | Protein Abundance & PTMs | LC-MS/MS, TMT/SILAC | Provides direct enzyme abundance data for more accurate constraint of maximal reaction fluxes (Vmax). | ~10,000-15,000 proteins |
| Metabolomics | Metabolite Concentration | GC/LC-MS, NMR | Provides extracellular exchange rates and intracellular concentration data for thermodynamic (e.g., Gibbs) constraints. | 1,000-5,000 metabolites |
Detailed Protocols for Omics-Guided FBA
Protocol 2.1: Generating Transcriptomics Data for Model Constraint (RNA-Seq) Objective: Generate gene expression data to create a context-specific metabolic model from a generic genome-scale reconstruction (GEM).
Sample Preparation & Sequencing:
- Extract total RNA from target cells/tissue using a TRIzol-based kit. Assess integrity (RIN > 8).
- Prepare poly-A enriched libraries using a stranded mRNA kit (e.g., Illumina TruSeq).
- Sequence on an Illumina NovaSeq platform to a depth of 30-50 million 150bp paired-end reads per sample.
Bioinformatic Processing:
- Quality Control: Use FastQC and Trimmomatic to assess and trim adapter/low-quality sequences.
- Alignment & Quantification: Align reads to the reference genome (e.g., GRCh38) using STAR aligner. Quantify gene-level counts using featureCounts.
- Normalization: Convert raw counts to Transcripts Per Million (TPM) using a custom script:
TPM = (Reads per Gene * 10^6) / (Gene Length * Sum(Reads/Gene Length)).
Integration with GEM:
- Map gene identifiers from RNA-Seq to gene identifiers in the metabolic model (e.g., Recon3D).
- Apply an expression threshold (e.g., TPM > 1) to determine "present" genes. Reactions are only included in the context-specific model if >50% of their associated genes (via Gene-Protein-Reaction rules) are present.
- Use the tINIT or FastCore algorithm (in COBRA Toolbox for MATLAB/Python) to generate the tissue-specific model.
Protocol 2.2: LC-MS/MS Metabolomics for Exchange Flux Determination Objective: Quantify extracellular metabolite uptake/secretion rates to constrain the exchange reaction bounds in an FBA model.
Experimental Setup & Quenching:
- Culture cells in a defined medium. Collect supernatant samples at multiple timepoints during exponential growth.
- Immediately quench metabolism by mixing 1 mL of supernatant with 4 mL of cold (-40°C) 100% methanol. Centrifuge (15,000 x g, 10 min, -9°C).
Sample Analysis via LC-MS:
- Reconstitute dried extracts in 100 µL of LC-MS grade water.
- Inject 5 µL onto a HILIC column (e.g., SeQuant ZIC-pHILIC) coupled to a high-resolution mass spectrometer (e.g., Q Exactive HF).
- Run a gradient from 80% acetonitrile/20% 20mM ammonium carbonate (pH 9.2) to 20% acetonitrile over 15 min.
- Acquire data in full-scan, negative ion mode (m/z 70-1000) at 120,000 resolution.
Data Processing & Flux Calculation:
- Process raw files using MS-DIAL or XCMS for peak picking, alignment, and annotation against an in-house standard library.
- Quantify concentration changes over time using internal standards (e.g., 13C-labeled amino acids).
- Calculate specific uptake/secretion rates (in mmol/gDW/h) by fitting to the equation:
dC/dt = q * X, where C is concentration, q is specific rate, and X is biomass concentration.
Visualization of the Integrative Omics-to-FBA Workflow
Title: Multi-Omics Data Integration for FBA Workflow
Title: Omics Constraints Directing FBA Flux Predictions
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents & Kits for Multi-Omics Sample Preparation
| Reagent/Kits | Provider (Example) | Function in Workflow |
|---|---|---|
| TRIzol Reagent | Thermo Fisher Scientific | Simultaneous extraction of high-quality RNA, DNA, and proteins from a single sample for multi-omics studies. |
| TruSeq Stranded mRNA Kit | Illumina | Library preparation for RNA-Seq, preserving strand information for accurate transcript quantification. |
| Tandem Mass Tag (TMT) 16plex | Thermo Fisher Scientific | Enables multiplexed quantitative proteomics of up to 16 samples in a single LC-MS/MS run, improving throughput and reducing variance. |
| SeQuant ZIC-pHILIC HPLC Column | MilliporeSigma | Stationary phase for hydrophilic interaction liquid chromatography (HILIC), essential for separating polar metabolites in LC-MS metabolomics. |
| C13 Labeled Internal Standard Mix | Cambridge Isotope Labs | Mixture of uniformly labeled metabolites for absolute quantification and correction of matrix effects in mass spectrometry-based metabolomics. |
| COBRA Toolbox | Open Source (GitHub) | MATLAB/Python suite for constraint-based reconstruction and analysis, containing algorithms (tINIT, FASTCORE) for integrating omics data. |
Why Integrate? The Power of Data-Driven Constraint for Metabolic Predictions
Integrating diverse omics datasets (genomics, transcriptomics, proteomics, metabolomics) with constraint-based metabolic models, such as those analyzed through Flux Balance Analysis (FBA), addresses a core limitation: the underdetermination of metabolic flux states. Genome-scale models (GEMs) define a vast solution space of possible flux distributions. Data-driven constraints, derived from experimental omics measurements, rigorously narrow this space, leading to more physiologically accurate and context-specific predictions. This integration is essential for applications in metabolic engineering, identification of drug targets in pathogens or cancer, and understanding of metabolic adaptations in disease.
Key Quantitative Data on Integration Impact
Table 1: Impact of Omics Data Integration on Model Prediction Accuracy
| Omics Data Type | Constraint Method | Typical Reduction in Solution Space | Reported Improvement in Prediction vs. Experimental Validation | Key Reference (Example) |
|---|---|---|---|---|
| Transcriptomics | GENE-Protein-Reaction (GPR) rules + Expression thresholds (e.g., iMAT, INIT) | 40-70% | Flux predictions: R² improvement from ~0.3 to ~0.6-0.8 | Machado et al., 2016 |
| Proteomics | Direct enzyme abundance constraints (E-Flux) | 30-60% | Growth rate prediction error reduced by up to 50% | Becker & Palsson, 2008 |
| Metabolomics | Thermodynamic (Loopless) constraints + Concentration-derived flux bounds | 20-50% | Correct prediction of futile cycle directionality >90% | Henry et al., 2007 |
| 13C-Fluxomics | Direct fluxomic constraints for key central carbon metabolism nodes | 60-90% | Central carbon flux correlation R² > 0.9 | Sauer et al., 1999 |
| Multi-omics (e.g., transcript + protein) | Integrative algorithms (e.g., METRADE, GECKO) | 70-85% | Context-specific model extraction accuracy >85% | Sánchez et al., 2017 |
Table 2: Comparison of Major Data Integration Algorithms
| Algorithm Name | Primary Data Inputs | Constraint Principle | Software/Toolbox | Best For |
|---|---|---|---|---|
| iMAT | Transcriptomics | Categorizes reactions as High/Low/Medium activity; maximizes flux of active reactions. | COBRA Toolbox | Tissue/cell-specific model reconstruction. |
| E-Flux | Transcriptomics/Proteomics | Treats expression data as proportional to maximum reaction capacity (Vmax). | COBRA Toolbox | Condition-specific flux predictions. |
| GECKO | Proteomics | Incorporates enzyme kinetics and abundance into GEMs as explicit constraints. | GECKO Toolbox | Resource balance analysis, predicting enzyme limitations. |
| OMNI | Multi-omics (Geno, Trans, Proteo, Metabo) | Probabilistic integration using Bayesian inference to weight data sources. | -- | Integrative analysis of heterogeneous datasets. |
| REM | Metabolomics | Uses exo-/endometabolome data to fit a thermodynamically feasible flux profile. | -- | Thermodynamics-aware flux estimation. |
Experimental Protocols
Protocol 3.1: Generating a Context-Specific Model Using Transcriptomic Data and iMAT
Objective: Reconstruct a cancer cell line-specific metabolic model from a generic human GEM (e.g., Recon3D) and RNA-Seq data.
Materials: High-quality RNA-Seq data (FPKM/TPM values) for target cell line, reference human GEM, COBRA Toolbox for MATLAB/Python.
Procedure:
- Data Preprocessing: Normalize RNA-Seq data (e.g., TPM). Map gene identifiers to those in the GEM's GPR associations.
- Discretization: For each gene, discretize expression into three states: High (top percentile, e.g., >75th), Low (bottom percentile, e.g., <25th), and Medium.
- Reaction Assignment: Using GPR rules, assign each reaction a state:
- If all associated genes are High → reaction is active.
- If all associated genes are Low → reaction is inactive.
- Otherwise → reaction is medium.
- iMAT Optimization: Formulate and solve a mixed-integer linear programming (MILP) problem that:
- Maximizes the sum of fluxes through reactions labeled active.
- Minimizes the sum of fluxes through reactions labeled inactive.
- Subject to the stoichiometric constraints (S*v = 0) and standard flux bounds.
- Model Extraction: Extract the consistent, active subnetwork from the iMAT solution to create the context-specific model.
- Validation: Simulate known metabolic phenotypes (e.g., essentiality of glycolysis) and compare predictions to independent experimental data (e.g., CRISPR screen gene essentiality).
Protocol 3.2: Constraining Models with Absolute Proteomics Data Using GECKO
Objective: Enhance a yeast GEM (e.g., Yeast8) with measured enzyme abundances to predict growth under different nutrient conditions.
Materials: Absolute protein abundance data (mg protein/gDW), genome-scale enzyme-constrained model (ecYeast), GECKO toolbox.
Procedure:
- Enhance GEM: Use the
enhanceGEMfunction in GECKO to add pseudo-metabolites (representing enzymes) and pseudo-reactions (representing enzyme usage) to the base GEM. - Apply Proteomic Constraints: Incorporate measured enzyme abundances as upper bounds for the corresponding enzyme usage reactions.
- Define Kinetic Parameters: Assign approximate kcat values (from databases like BRENDA) to each enzyme-reaction pair.
- Resource Balance Analysis: Formulate an optimization problem where the objective (e.g., growth) is maximized subject to stoichiometry, kinetic (
enzyme * kcat ≥ flux), and total enzyme pool constraints. - Simulation & Prediction: Predict growth rates and flux distributions under different media. The model will naturally re-allocate enzyme resources based on optimization.
- Analysis: Identify enzyme-limited reactions and compare predicted vs. measured growth phenotypes.
Mandatory Visualizations
Title: Omics Data Integration Workflow for Constraining Metabolic Models
Title: Sequential Reduction of Metabolic Solution Space via Omics Constraints
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Data-Driven Constraint Studies
| Item | Function/Application in Integration Protocols | Example Product/Resource |
|---|---|---|
| Genome-Scale Metabolic Models (GEMs) | The foundational stoichiometric matrix of reactions and metabolites for constraint-based analysis. | Human: Recon3D, HMR; Yeast: Yeast8; E. coli: iML1515; Generic: ModelSEED. |
| COBRA Toolbox | Primary software suite (MATLAB/Python) for performing FBA and implementing integration algorithms (iMAT, E-Flux). | https://opencobra.github.io/cobratoolbox/ |
| GECKO Toolbox | Specialized toolbox for enhancing GEMs with enzyme constraints using proteomic data. | https://github.com/SysBioChalmers/GECKO |
| RNA-Seq Analysis Pipeline | For quantifying gene expression from raw sequencing reads (FASTQ) to model-compatible values (TPM). | Tools: STAR (alignment), featureCounts/HTSeq (quantification), DESeq2/edgeR (normalization). |
| LC-MS/MS Platform for Proteomics | For generating absolute or relative protein abundance data to constrain enzyme capacity. | Platforms: Thermo Orbitrap, SCIEX TripleTOF. Software: MaxQuant, Proteome Discoverer. |
| Mass Spectrometry for Metabolomics | For quantifying intracellular and extracellular metabolite concentrations. | GC-MS (for polar metabolites), LC-MS (broad coverage). Software: XCMS, MS-DIAL. |
| 13C-Labeled Substrates | Essential for conducting fluxomics experiments to determine in vivo metabolic flux rates. | [1,2-13C]Glucose, [U-13C]Glucose, 13C-Glutamine. |
| Constraint-Solving Optimizer | Solver for the linear (LP) and mixed-integer (MILP) problems generated during integration. | Gurobi, CPLEX, IBM ILOG (commercial); GLPK, SCIP (open-source). |
| Omics Data Mapping Database | Provides consistent identifiers to map genes, proteins, and metabolites between datasets and the GEM. | UniProt (proteins), HMDB (metabolites), KEGG/ModelSEED (reactions). |
Application Notes
The integration of multi-omics data with Flux Balance Analysis (FBA) is pivotal for constructing genome-scale metabolic models (GEMs) that accurately reflect the physiological state of a specific cell, tissue, or disease context. Two primary paradigms govern this integration: Top-Down and Bottom-Up reconstruction.
Top-Down Approach: Begins with an existing, generic, genome-scale metabolic reconstruction. This generic model is then systematically constrained and refined using context-specific omics data (e.g., transcriptomics, proteomics) to eliminate inactive reactions and pathways, yielding a cell-type or condition-specific model. It is efficient and leverages prior knowledge but may be biased by the starting model's composition.
Bottom-Up Approach: Starts de novo from a curated set of metabolic functions known to be present in the specific context, often derived from omics data and literature. This core model is then expanded iteratively. It minimizes bias from generic models but is labor-intensive and may miss peripheral pathways.
The choice of paradigm depends on the research goal, data availability, and desired model specificity. Top-down is favored for high-throughput generation of context-specific models across conditions or cell types. Bottom-up is essential for modeling poorly characterized systems or when maximum biochemical accuracy for a core process is required.
Protocols
Protocol 1: Top-Down Reconstruction via FASTCORE Objective: Generate a context-specific metabolic model from a generic GEM using transcriptomic data. Materials: Generic GEM (e.g., Recon3D, Human1), transcriptomics data (RPKM/TPM values), COBRA Toolbox in MATLAB/Python. Procedure:
- Data Preprocessing: Map transcriptomic features to model gene identifiers. Define a present/absent call (e.g., transcripts per million (TPM) > 1).
- Generate Context-Specific Reaction List: Identify reactions where all associated genes are called "present" (logical AND) or at least one is present (logical OR), depending on the desired stringency.
- Apply FASTCORE Algorithm:
- Input the generic model (
S matrix,lb,ub) and the list of "core" reactions presumed active. - FASTCORE finds a flux-consistent subnetwork of the generic model that contains all core reactions while maintaining network connectivity.
- It solves a series of linear programming (LP) problems to minimize the number of supporting reactions added.
- Input the generic model (
- Model Extraction & Validation: Extract the subnetwork as the new context-specific model. Test for basic functionality (e.g., ATP production, biomass synthesis) and compare predicted essential genes with experimental knockdown data.
Protocol 2: Bottom-Up Reconstruction for a Metabolic Subsystem Objective: Construct a core model of mitochondrial fatty acid oxidation (FAO) de novo. Materials: Genome annotation, proteomics data for mitochondrial proteins, biochemical literature (e.g., BRENDA), pathway databases (MetaCyc), modelling environment (e.g., PySCeS, COBRApy). Procedure:
- Define System Boundary: Specify the subsystem (e.g., mitochondrial FAO from C4 to C16 acyl-CoAs), the cellular compartment, and input/output metabolites (e.g., Acyl-CoA, NAD+, FAD, Acetyl-CoA, NADH, FADH2).
- Curate Reaction List: Using proteomic data and literature, list all enzymatic reactions. For each, record: EC number, stoichiometry, reversibility, gene-protein-reaction (GPR) rules, and subcellular location.
- Assemble Stoichiometric Matrix (S): Compile reactions into the
Smatrix where rows are metabolites and columns are reactions. - Add Transport and Demand Reactions: Include reactions for substrate uptake, product secretion, and a "demand" for ATP synthesis linked to the electron transport chain.
- Define Constraints: Set lower (
lb) and upper (ub) flux bounds based on reversibility (e.g., 0 to 1000 for irreversible, -1000 to 1000 for reversible). Apply capacity constraints if kinetic data is available. - Model Testing & Gap-Filling: Perform flux variability analysis (FVA). Test if the network can produce expected outputs. If gaps exist, iteratively consult literature and data to add missing reactions and curate GPR rules.
Data Presentation
Table 1: Comparative Analysis of Top-Down vs. Bottom-Up Reconstruction Paradigms
| Feature | Top-Down Paradigm | Bottom-Up Paradigm |
|---|---|---|
| Starting Point | Existing generic GEM | Omics data & biochemical literature |
| Core Methodology | Constraint-based pruning (e.g., FASTCORE, INIT) | De novo biochemical assembly |
| Primary Omics Data | Transcriptomics, Proteomics | Proteomics, Literature curation |
| Computational Speed | Fast (minutes-hours) | Slow (weeks-months) |
| Risk of Bias | High (inherited from generic model) | Low |
| Coverage | Broad, genome-scale | Narrow, subsystem-focused |
| Key Output | Context-specific GEM | High-confidence core model |
| Best For | Multi-condition comparisons, high-throughput studies | Novel pathways, high biochemical accuracy |
Table 2: Example Flux Comparison: Generic vs. Hepatocyte-Specific Model (Top-Down)
| Metabolic Function | Generic Liver GEM Flux (mmol/gDW/h) | Hepatocyte-Specific Model Flux (mmol/gDW/h) | Data Source for Constraint |
|---|---|---|---|
| Albumin Synthesis | 0.001 - 0.1 | 0.05 | Proteomics (He et al., 2020) |
| Urea Cycle | 0.1 - 20 | 15.2 | Transcriptomics (GTEx, 2023) |
| Glycolysis | 0 - 50 | 8.7 | Transcriptomics (GTEx, 2023) |
| CYP450 Metabolism | 0 - 5 | 3.1 | Proteomics (He et al., 2020) |
Mandatory Visualizations
Title: Top-Down Model Reconstruction Workflow
Title: Bottom-Up Core Model Assembly Process
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Context-Specific Model Reconstruction
| Item / Solution | Function in Research | Example |
|---|---|---|
| Generic Metabolic Reconstructions | High-quality starting point for top-down reconstruction. Provides comprehensive biochemical network. | Recon3D, Human1, HMR, AGORA |
| COBRA Toolbox | Primary computational platform for constraint-based modeling, containing implementation of key algorithms. | fastCore(), init() functions. |
| Omics Data Repositories | Source of context-specific transcriptomic/proteomic data to constrain models. | GTEx Portal, Human Protein Atlas, GEO, PRIDE. |
| Biochemical Pathway Databases | Reference for reaction stoichiometry, EC numbers, and metabolite IDs during bottom-up curation. | MetaCyc, BRENDA, Rhea, KEGG. |
| Metabolite & Reaction Identifier Mappers | Crucial for harmonizing identifiers between omics datasets and model components. | MetaboAnalyst, BridgeDb, chemCompID mapping files. |
| Gene Essentiality Datasets | Used for validating the predictive capability of the reconstructed context-specific model. | CRISPR screens (DepMap), siRNA databases. |
| High-Performance Computing (HPC) Cluster | Enables large-scale sampling and analysis of genome-scale models, especially for multi-condition studies. | Slurm-managed clusters, cloud computing (AWS, GCP). |
A Step-by-Step Pipeline: Methodologies and Cutting-Edge Applications in Biomedicine
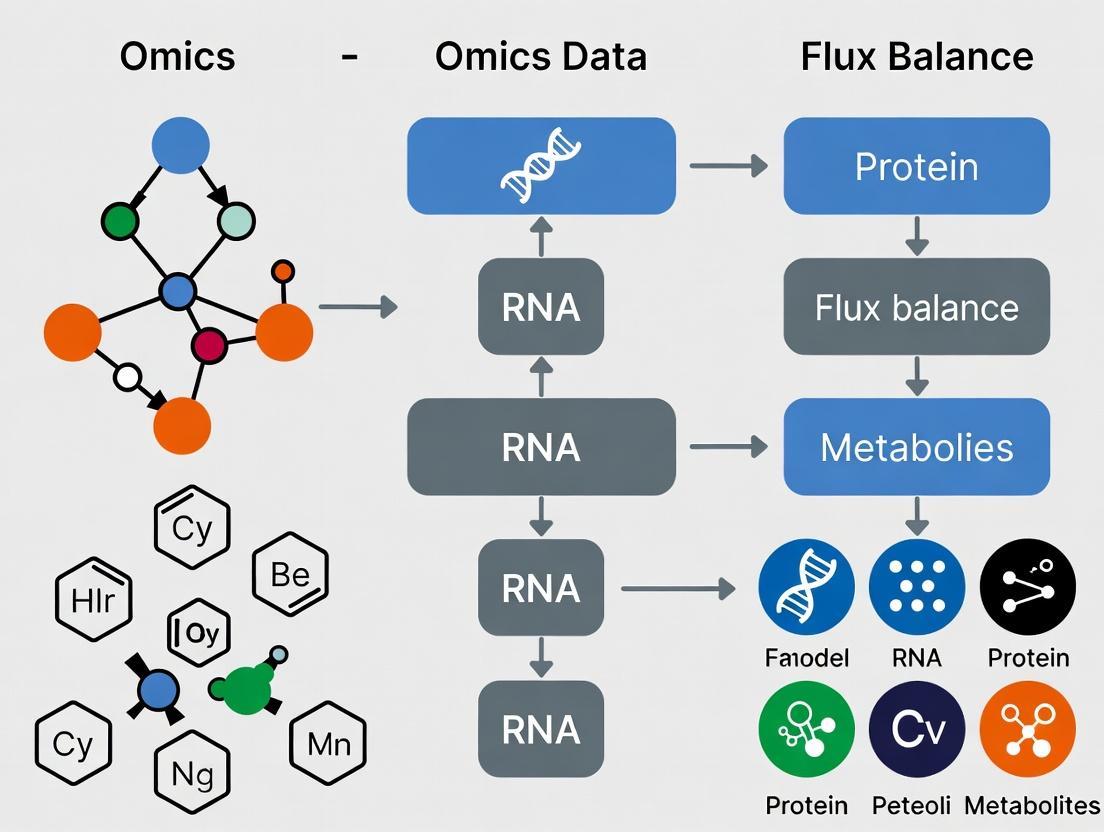
The integration of transcriptomics, proteomics, and metabolomics data with Flux Balance Analysis (FBA) provides a powerful framework for constructing genome-scale metabolic models (GEMs) that reflect specific physiological states. This protocol details standardized procedures for acquiring and preprocessing multi-omics inputs to generate quantitative constraints for FBA, a core component of thesis research on integrated multi-omics and metabolic modeling.
Table 1: Common Multi-Omics Platforms and Output Characteristics
| Omics Layer | Primary Platform | Typical Output Format | Key Preprocessing Metric | FBA-Relevant Conversion |
|---|---|---|---|---|
| Transcriptomics | RNA-Seq (Illumina) | FASTQ -> Count Matrix | TPM (Transcripts Per Million) | Relative enzyme level proxies (via GPR rules). |
| Proteomics | LC-MS/MS (TMT/Isobaric) | Raw Spectra -> Peptide Intensities | LFQ (Label-Free Quantification) Intensity | Absolute or relative enzyme abundance constraints. |
| Metabolomics | GC-MS / LC-MS | Peak Areas -> Compound Intensities | Peak Area, Normalized to internal standard | Extracellular exchange or internal flux bounds. |
Table 2: Standardization Parameters for FBA Integration
| Processing Step | Transcriptomics | Proteomics | Metabolomics |
|---|---|---|---|
| Normalization | DESeq2 (Median of Ratios) / TPM | Cyclic LOESS (vs. reference channel) | Probabilistic Quotient Normalization (PQN) |
| Imputation (Missing Data) | Not applicable (zero count = no expression) | Minimum value imputation (MNAR assumption) | K-Nearest Neighbors (KNN) imputation |
| Scaling | Log2(TPM + 1) | Log2(LFQ intensity) | Autoscaling (mean-centered, unit variance) |
| FBA Mapping | Map to genes in GEM via Gene-Protein-Reaction (GPR) Boolean rules. | Map directly to enzyme subunits in GEM. | Map KEGG/Model SEED IDs to model metabolite IDs. |
Experimental Protocols
Protocol 3.1: RNA-Seq Data Processing for Transcriptomic Constraints Objective: Generate gene expression proxies for reaction weights in FBA.
- Quality Control: Use
FastQCon raw FASTQ files. Trim adapters and low-quality bases withTrim Galore!(default parameters). - Alignment & Quantification: Align reads to the reference genome using
STAR(--quantMode GeneCounts). Use the corresponding genome annotation file (GTF). - Normalization: Load gene count matrix into R/Bioconductor. Use
DESeq2to calculate size factors and generate normalized counts. Convert to TPM using gene lengths. - Mapping to Metabolic Model: a. Ensure gene identifiers (e.g., Ensembl IDs) match those in your GEM's GPR rules. b. For each reaction, evaluate its GPR rule (e.g., "gene1 AND gene2") using the normalized expression vector. A common method is to assign the reaction value as the minimum expression of genes in an AND clause and the maximum for an OR clause. c. Create a reaction abundance vector for use in methods like E-Flux or GECKO.
Protocol 3.2: LC-MS Proteomics Data Preprocessing Objective: Obtain quantitative protein abundances for direct enzyme constraint.
- Raw Data Processing: Process .raw files using
MaxQuantorDIA-NN.- Search Parameters: Set fixed (e.g., Cysteine alkylation) and variable modifications (e.g., Methionine oxidation). Use the appropriate organism-specific FASTA database.
- Quantification: For TMT, select "Reporter ion MS2" (or MS3). For LFQ, enable the LFQ algorithm.
- Post-Processing in R:
a. Load the
proteinGroups.txt(MaxQuant) output. Filter: Remove reverse hits, contaminants, and proteins only identified by site. b. Normalization (TMT): Uselimma'snormalizeCyclicLoessfunction on log2-transformed reporter ion intensities. c. Normalization (LFQ): Use the provided LFQ intensities. Perform median normalization on the log2 intensities. d. Imputation: For missing values assumed to be missing not at random (MNAR), impute using a constant low value (e.g., distribution down-shift of 1.8 SDs). - Model Mapping: Map UniProt IDs to model enzyme identifiers. Use abundance values directly to constrain enzyme capacity in the GEM.
Protocol 3.3: Targeted Metabolomics Data Standardization for Exchange Fluxes Objective: Generate absolute quantitative extracellular metabolite data to set realistic exchange flux bounds in FBA.
- Peak Integration & Calibration: Use vendor software (e.g., Agilent MassHunter) or
XDATAto integrate peaks for target metabolites. Generate a calibration curve (peak area vs. known concentration) for each. - Concentration Calculation: Calculate extracellular concentrations (µM or mM) in the media from peak areas using the linear fit from the calibration curve.
- Unit Conversion to Flux Bounds:
a. Convert concentration [C] to amount using culture volume.
b. Calculate the maximum uptake or secretion rate:
v_max = (Amount) / (Cell Count * Time). c. Set the lower bound (lb) for the exchange reaction in the model. Example: For a consumed metabolite, setlb = -v_max. For a secreted one, set the upper bound (ub) =v_max.
Visualization of Workflows and Relationships
(Multi-Omics Data Processing for FBA)
(Multi-Omics Data Integration with FBA)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Multi-Omics Sample Preparation
| Item | Function/Description | Example Product/Catalog |
|---|---|---|
| TriZol/ TRI Reagent | Simultaneous extraction of RNA, DNA, and protein from a single biological sample. Essential for paired omics from limited material. | Invitrogen TRIzol Reagent (15596026) |
| Phase Lock Gel Tubes | Facilitates clean separation of organic and aqueous phases during TRIzol extraction, improving RNA yield and purity. | Quantabio Phase Lock Gel Heavy (2302830) |
| Protease & Phosphatase Inhibitor Cocktails | Added to lysis buffers to prevent protein degradation and preserve post-translational modification states during proteomics prep. | Thermo Scientific Halt Cocktail (78442) |
| Mass-Spec Grade Trypsin/Lys-C Mix | High-purity enzymes for reproducible and complete protein digestion into peptides for LC-MS/MS analysis. | Promega Trypsin/Lys-C Mix (V5073) |
| TMTpro 16plex Kit | Isobaric labeling reagents for multiplexed quantitative proteomics, allowing parallel analysis of up to 16 samples in one MS run. | Thermo Scientific TMTpro 16plex (A44522) |
| Stable Isotope-Labeled Internal Standards | Absolute quantification standards for targeted metabolomics (e.g., 13C, 15N-labeled amino acids, nucleotides). | Cambridge Isotope Laboratories (Various) |
| Dual-Sequence Specific Indexed Adapters | For multiplexed RNA-Seq library prep, enabling pooling of samples and demultiplexing post-sequencing. | Illumina IDT for Illumina RNA UD Indexes |
| RNeasy Mini Kit / QIAprecipitate | For clean-up and concentration of RNA or metabolites after extraction, removing salts and contaminants. | Qiagen RNeasy Kit (74106) |
| BCA or Qubit Protein Assay Kits | Quantification of total protein concentration prior to proteomics workflow for equal loading. | Thermo Scientific Pierce BCA Assay Kit (23225) |
| SP3 Magnetic Beads | For detergent-free, scalable protein cleanup, digestion, and TMT labeling prior to LC-MS/MS. | Cytiva Sera-Mag SpeedBeads (45152105050250) |
In the context of a thesis on Integrating omics data with flux balance analysis (FBA) research, the reconciliation of high-throughput molecular data with genome-scale metabolic models (GEMs) is a central challenge. FBA provides a powerful constraint-based modeling framework but generates an in-silico metabolic network that may not reflect a specific cell's or tissue's actual, context-specific state. Omics data (transcriptomics, proteomics, metabolomics) offer this context but are not inherently mechanistic. The algorithms FASTCORE, GIMME, INIT, and CORDA are pivotal for building accurate, condition-specific metabolic models (CSMs) by systematically integrating omics data into GEMs, thereby enhancing predictive capabilities for biomedical and biotechnological applications.
Detailed Application Notes and Protocols
FASTCORE
Purpose: Generates a consistent, context-specific core model from a global GEM based on a set of high-confidence reactions (e.g., from highly expressed genes). Core Principle: Uses linear programming (LP) to find the minimum set of reactions from the global network that can carry flux on all "core" reactions, ensuring network connectivity and thermodynamic consistency. Typical Input: 1) Global GEM (e.g., Recon, Human1), 2) A binary vector or list specifying which reactions in the global model are part of the "core" set. Output: A pruned, functional core metabolic network model.
Protocol: Generating a Context-Specific Model with FASTCORE
- Prepare Omics Data: Process transcriptomic (RNA-seq/microarray) or proteomic data. Map gene/protein identifiers to model gene-reaction rules (GPRs). Define an expression threshold.
- Define Core Reaction Set: For each reaction, evaluate its associated GPR against the omics data. Mark a reaction as "core" if its associated genes are expressed above the threshold (e.g., TPM > 1, protein detected).
- Algorithm Execution: Use the FASTCORE algorithm (available in COBRApy or MATLAB COBRA Toolbox).
- Model Validation: Check for metabolic functionality (e.g., ability to produce essential biomass precursors) and compare predicted flux distributions against experimental data (e.g., growth rates, substrate uptake).
GIMME (Gene Inactivity Moderated by Metabolism and Expression)
Purpose: Creates a context-specific model by minimizing the flux through reactions associated with lowly expressed genes, subject to a defined metabolic objective (e.g., biomass production). Core Principle: Uses quadratic programming to minimize the weighted sum of fluxes through "inactive" reactions while requiring a minimum objective function flux. Typical Input: 1) Global GEM, 2) Gene expression data mapped to the model, 3) A threshold for "low" expression, 4) A required minimum flux for a biological objective (e.g., 10% of optimal growth). Output: A functional CSM with penalized low-expression reaction fluxes.
Protocol: Integrating Expression Data with GIMME
- Data Mapping & Normalization: Normalize gene expression data (e.g., RPKM/TPM to Z-scores). Map genes to reactions via GPRs using Boolean logic (AND/OR).
- Assign Reaction Scores: For each reaction, assign a score based on the expression of its associated gene(s). For example, use the maximum expression value for genes linked by OR, and the minimum for genes linked by AND.
- Define Parameters: Set an expression threshold to distinguish active/inactive genes. Define the metabolic objective (e.g.,
biomass_reaction) and its minimum required flux (obj_frac, e.g., 0.1 for 10% of maximal). - Run GIMME Optimization: Solve the quadratic minimization problem.
- Analyze Results: Identify reactions forced to carry zero flux due to low expression. Validate model predictions against known metabolic phenotypes.
INIT (Integrative Network Inference for Tissues)
Purpose: Generates a tissue-specific model by integrating quantitative transcriptomic and proteomic data as well as metabolomic and literature-based evidence (e.g., from HPA) to define reaction confidence scores. Core Principle: Uses linear programming to find the model with the maximum total confidence score (sum of weights for included reactions) that can produce a set of known metabolic functions (e.g., secrete specific metabolites). Typical Input: 1) Global GEM, 2) Quantitative omics data (RNA-seq, proteomics), 3) Metabolomic data (e.g., from HMDB) defining a set of "core" metabolites that must be produced/consumed, 4) Literature-based evidence. Output: A quantitative, functional tissue-specific metabolic model.
Protocol: Building a Tissue-Specific Model with INIT
- Compile Evidence & Calculate Weights: For each reaction i, compile evidence from:
- Transcriptomics (mRNA level)
- Proteomics (protein abundance)
- Metabolomics (substrate/product presence)
- Literature (manual curation)
Integrate these into a single quantitative weight (
w_i) using a scoring function.
- Define Metabolite Core Set: From metabolomic databases (e.g., HMDB) and literature, define a set of metabolites (
M_core) that are known to be produced or consumed by the target tissue/cell type. - Run INIT Linear Programming:
- Objective: Maximize ∑(wi * yi), where
y_iis a binary variable indicating inclusion of reaction i. - Constraints: 1) The network must be able to produce/consume all metabolites in
M_coreat a non-zero rate. 2) The network must be stoichiometrically balanced.
- Objective: Maximize ∑(wi * yi), where
- Implementation: Use the
createTissueSpecificModelfunction in the RAVEN Toolbox for MATLAB, which implements INIT.
CORDA (Cost Optimization Reaction Dependency Assessment)
Purpose: Generates high-quality CSMs by using omics data to classify reactions into sets of high-confidence (HC), medium-confidence (MC), and low-confidence (LC) based on multiple evidence sources, then optimizes for a minimal network satisfying all HC and a subset of MC reactions. Core Principle: Uses mixed-integer linear programming (MILP) to find the network that includes all HC reactions, excludes all LC reactions, and includes a maximal weighted sum of MC reactions, while maintaining network functionality. Typical Input: 1) Global GEM, 2) Gene/protein expression data, 3) Manual curation inputs to classify reactions into HC, MC, LC sets. Output: A high-confidence, functional CSM.
Protocol: High-Confidence Model Reconstruction with CORDA
- Reaction Confidence Scoring: For each reaction, assign a confidence score (e.g., 0: LC, 1: MC, 2: HC) based on integrated evidence. This can be automated via expression thresholds but often involves manual curation for HC/LC sets.
- Define Metabolic Tasks: Define a set of metabolic functions (
tasks) the final model must perform (e.g., ATP production, lipid synthesis, known secretion products). These are used as functional constraints. - Run CORDA Optimization: The MILP formulation:
- Variables: Binary variable (
v_i) for inclusion of each reaction i. - Objective: Maximize ∑(weightMCi * v_i) for MC reactions.
- Constraints:
v_i = 1for all HC reactions (mandatory).v_i = 0for all LC reactions (forbidden).- The network must satisfy all defined metabolic
tasks. - Stoichiometric consistency (mass balance).
- Variables: Binary variable (
- Implementation: Available as a standalone MATLAB function or within the CORDA Python package.
Table 1: Comparative Overview of Core Algorithms for Omics-FBA Integration
| Algorithm | Core Mathematical Method | Primary Omics Input | Key Strength | Key Limitation | Typical Output Size (% of Global Model) |
|---|---|---|---|---|---|
| FASTCORE | Linear Programming (LP) | Binary reaction activity (from transcript./proteom.) | Fast, ensures a consistent, connected core model. | Relies on a binary core set; may include non-expressed reactions for connectivity. | 20-40% |
| GIMME | Quadratic Programming (QP) | Continuous gene expression values | Minimizes usage of low-expression reactions; maintains defined metabolic objective. | Requires user-defined expression threshold and objective fraction. | 30-60% |
| INIT | Linear Programming (LP) | Quantitative multi-omics (transcript., proteom., metabolom.) | Integrates multiple data types quantitatively; maximizes total evidence score. | Complex weight calculation; requires a predefined set of core metabolites. | 40-70% |
| CORDA | Mixed-Integer LP (MILP) | Reaction confidence scores (-1,0,1) | High flexibility with HC/MC/LC classification; produces high-confidence models. | Computationally intensive; manual curation often needed for confidence scoring. | 15-50% |
Visualizations
Workflow for Integrating Omics Data with GEMs using Core Algorithms
GIMME vs CORDA: Algorithmic Approach Comparison
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Omics-FBA Integration Studies
| Item / Reagent | Provider / Example | Function in Protocol |
|---|---|---|
| Reference Genome-Scale Model (GEM) | Human1, Recon3D, HMR, iJO1366 (E. coli) | The foundational, organism-specific metabolic network used as a template for all context-specific reconstructions. |
| High-Throughput Omics Data | RNA-seq data (Illumina), LC-MS/MS Proteomics (Thermo Fisher) | Provides the condition- or tissue-specific molecular readouts (gene/protein expression) used to constrain the global model. |
| Metabolomic Database | Human Metabolome Database (HMDB), Yeast Metabolome Database (YMDB) | Source of evidence for metabolite presence/absence, used to define core metabolic tasks (especially for INIT). |
| Modeling & Algorithm Software | COBRA Toolbox (MATLAB), COBRApy (Python), RAVEN Toolbox (MATLAB) | Software suites containing implementations of FASTCORE, GIMME, INIT, and other essential algorithms for constraint-based modeling. |
| Linear/Quadratic Programming Solver | Gurobi, CPLEX, IBM ILOG | Commercial optimization solvers (often required for MILP problems in CORDA) to compute model solutions efficiently. |
| Curation Database | UniProt, PubMed, BRENDA | Resources for manual curation of reaction evidence, gene-protein-reaction (GPR) rules, and confidence scoring. |
| Validation Data Set | Extracellular flux data (Seahorse Analyzer, Agilent), SILAC fluxomics | Experimental data on metabolic uptake/secretion or intracellular fluxes used to validate predictions of the generated CSM. |
Integrating transcriptomic data with Genome-Scale Metabolic Models (GEMs) enables the creation of context-specific metabolic networks, crucial for understanding tissue-specific physiology and disease mechanisms. This protocol details the generation of tissue- and condition-specific models using constraint-based reconstruction and analysis (COBRA) methods, directly supporting drug target identification and personalized medicine approaches within omics-integrated flux balance analysis (FBA) research.
The broader thesis of integrating multi-omics data with FBA aims to build predictive in silico models of cellular metabolism. Transcriptomics provides a key layer of information to constrain universal GEMs, such as Recon3D or HMR, to reflect the metabolic activity of a specific tissue (e.g., liver, heart, cancer) under defined conditions (e.g., normoxia, disease state). This process transforms a generic metabolic network into a functional model that can simulate condition-specific fluxes, predict essential genes, and identify therapeutic targets.
Application Notes: Core Methodologies & Data Comparison
Three primary algorithms are used for generating context-specific models. Their characteristics and data requirements are summarized below.
Table 1: Comparison of Major Context-Specific Model Reconstruction Algorithms
| Algorithm | Core Principle | Data Input Requirement | Key Strength | Primary Limitation |
|---|---|---|---|---|
| GIMME (Gene Inactivity Moderated by Metabolism and Expression) | Minimizes flux through reactions associated with low-expression genes while maintaining a predefined objective function (e.g., biomass). | Transcriptomic data (RPKM, TPM); Threshold for "low expression"; Reference GEM. | Robust, allows some low-expression activity if needed for network functionality. | Requires a user-defined expression threshold and objective function. |
| iMAT (Integrative Metabolic Analysis Tool) | Maximizes the number of high-expression reactions set to be active and low-expression reactions set to be inactive, satisfying stoichiometric constraints. | Transcriptomic data; High/Low expression thresholds; Reference GEM. | Does not require a predefined objective function; better for non-proliferating cells. | Computationally intensive; sensitive to threshold settings. |
| FastCore | Identifies a minimal consistent network from a reference GEM that contains a core set of reactions (e.g., those associated with high-expression genes). | A core set of reactions (e.g., from highly expressed genes); Reference GEM. | Fast, deterministic, and does not require expression thresholds or an objective. | Requires a predefined high-confidence core reaction set as input. |
Table 2: Typical Quantitative Output Metrics from Model Generation
| Metric | Description | Typical Range/Value (Example: Liver-Specific Model) |
|---|---|---|
| Model Reactions | Number of active reactions retained from the reference GEM. | 3,500 - 5,000 (from ~13,000 in Recon3D) |
| Model Genes | Number of associated genes retained. | 1,500 - 2,500 (from ~3,300 in Recon3D) |
| Functional Validation - ATP Max | Maximum achievable ATP synthesis flux (mmol/gDW/hr). | 5 - 15 |
| Functional Validation - Biomass | Production flux of a tissue-specific biomass reaction. | 0.01 - 0.1 (hr⁻¹) |
| Prediction Accuracy (vs. GRO data) | Correlation between predicted and experimental gene essentiality. | AUC: 0.65 - 0.85 |
Detailed Experimental Protocol: Generating a Tissue-Specific Model with iMAT
This protocol details the generation of a human cardiomyocyte-specific model from RNA-Seq data using the iMAT algorithm within the COBRA Toolbox for MATLAB.
Prerequisites and Software Setup
- Software: MATLAB, COBRA Toolbox (v3.0+), a solver (e.g., Gurobi, CPLEX).
- Reference GEM: Download Human-GEM (https://github.com/SysBioChalmers/Human-GEM) or Recon3D.
- Transcriptomic Data: RNA-Seq data (TPM or FPKM) from target tissue (e.g., GTEx portal: heart left ventricle) and a control/reference tissue.
Step-by-Step Procedure
Step 1: Data Preprocessing and Mapping
- Normalize Data: Ensure transcriptomic data is normalized (e.g., TPM). Log2-transform values.
- Define High/Low Thresholds: Calculate percentile thresholds. Common settings: High = 75th percentile, Low = 25th percentile of expression distribution across all samples.
- Map Genes to Model: Use the
mapExpressionToReactionsfunction to convert gene expression values to reaction scores, using GPR rules from the reference model.
Step 2: Prepare Inputs for iMAT
Step 3: Run iMAT Reconstruction
Step 4: Post-processing and Gap-Filling
- Check Connectivity: Ensure the model produces biomass precursors. Use
findBlockedReaction. - Perform Gap-Filling: Use the
fillGapsfunction to add minimal reactions from the reference model to allow basic functions (e.g., ATP maintenance, biomass production). - Add Tissue-Specific Constraints: Incorporate known nutrient uptake rates (from literature) using
changeRxnBounds.
Step 5: Validation and Analysis
- Test Basic Functionality: Simulate ATP maintenance (
DM_atp_c_) and tissue-relevant objective functions. - Compare with Experimental Data: Validate predicted essential genes against CRISPR screening data (e.g., DepMap) by performing in silico gene knockout simulations (
singleGeneDeletion). - Perform Flux Variability Analysis (FVA): Assess the flexibility of the network under different conditions.
Visualization of Workflows and Pathways
Title: Transcriptomics Integration Workflow for Tissue-Specific Models
Title: Omics-FBA Integration Loop for Prediction & Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for Transcriptomics-Integrated Metabolic Modeling
| Item / Resource | Function / Description | Example Source / Tool |
|---|---|---|
| Reference Genome-Scale Model (GEM) | A comprehensive, consensus metabolic network for the target organism. Serves as the template for reconstruction. | Human: Human1, Recon3D, HMR. Mouse: iMM1865. Generic: MetaCyc. |
| Curated Transcriptomic Datasets | High-quality, normalized gene expression data for the tissue/condition of interest. | GTEx Portal, ARCHS4, GEO, TCGA, ArrayExpress. |
| COBRA Toolbox | The standard MATLAB software suite for constraint-based modeling, containing all major reconstruction algorithms. | https://opencobra.github.io/cobratoolbox/ |
| Python cobrapy Package | Python implementation of COBRA methods, ideal for integration into larger bioinformatics pipelines. | https://cobrapy.readthedocs.io/ |
| Gurobi/CPLEX Optimizer | Commercial mathematical optimization solvers required for solving large linear programming problems in FBA. | Gurobi Optimization, IBM ILOG CPLEX. |
| Expression Mapping Tool | Software to accurately map gene IDs from expression data to model gene-protein-reaction (GPR) rules. | mapExpressionToReactions (COBRA), MERGE-Py. |
| Gene Essentiality Data | Experimental data for validating model predictions (e.g., CRISPR-Cas9 knockout screens). | DepMap Portal, OGEE, essentialgene.org. |
Incorporating Metabolomics and Proteomics for Enhanced Predictions
1.0 Application Notes
The integration of metabolomics and proteomics data with genome-scale metabolic models (GSMMs) through Flux Balance Analysis (FBA) represents a powerful paradigm for predictive systems biology. This multi-omics approach constrains the solution space of in silico models, transforming them from generic metabolic blueprints into condition-specific predictors of cellular phenotype. This is critical for applications in biotechnology and drug discovery, where accurate predictions of metabolic flux can identify novel drug targets or optimize bioproduction.
1.1 Key Advantages and Applications
- Target Identification: Proteomic constraints can pin-point over- or under-expressed enzymes, while metabolomic data can reveal accumulation or depletion of key metabolites. FBA simulations on this constrained model can predict which enzyme knockouts will lethally disrupt pathogen viability or cancer cell proliferation.
- Predicting Drug Mechanism of Action: Shifts in metabolite pools and enzyme abundance post-treatment can be integrated to model altered flux states. FBA can then infer the primary metabolic pathway inhibited, elucidating a drug's mechanism.
- Biomarker Discovery: Integrated models can simulate disease versus healthy states, predicting co-variation patterns between specific proteins and metabolites that serve as robust diagnostic or prognostic panels.
1.2 Quantitative Data Summary
Table 1: Comparison of Omics-Constraint Methods for FBA
| Constraint Type | Data Input | Typical FBA Integration Method | Key Effect on Model Prediction |
|---|---|---|---|
| Proteomics | Enzyme abundance (e.g., mg/gDW) | Thermodynamic (kcat) constraints via GECKO; Upper bound scaling via pFBA. | Reduces feasible flux space by limiting maximum turnover of reactions. Improves prediction of substrate uptake and growth rates. |
| Metabolomics | Metabolite concentration (e.g., µM) | Kinetic (Michaelis-Menten) constraints via MOMENT; Incorporation as inequality constraints. | Directs flux by defining metabolite availability. Can predict allosteric regulation points and pathway bottlenecks. |
| Multi-Omics | Combined protein & metabolite data | Steady-state modeling via METRADE or iterative fitting algorithms. | Maximizes consistency between all data layers and the metabolic network. Yields the most physiologically accurate flux distributions. |
Table 2: Impact of Omics Constraints on Model Accuracy (Representative Studies)
| Study Focus | Base Model Accuracy (R²) | Proteomics-Constrained Accuracy (R²) | Multi-Omics Constrained Accuracy (R²) | Key Prediction Validated |
|---|---|---|---|---|
| E. coli Growth Rate | 0.48 | 0.72 | 0.89 | Glucose uptake, acetate secretion |
| Cancer Cell Line (NCI-60) Proliferation | 0.31 | 0.65 | 0.78 | Essentiality of folate metabolism genes |
| S. cerevisiae Ethanol Production | 0.55 | 0.81 | 0.92 | Optimal oxygen uptake rate |
2.0 Experimental Protocols
2.1 Protocol: Generating Proteomics Data for GECKO Model Integration This protocol details the generation of absolute quantitative proteomics data suitable for constraining GSMMs using the GECKO toolbox.
Materials:
- Cell culture from condition of interest.
- Lysis buffer (e.g., 8M Urea, 50mM Tris-HCl pH 8.0).
- Protease/Phosphatase inhibitors.
- BCA Assay Kit.
- Trypsin/Lys-C protease mix.
- Stable Isotope Labeled Standard peptides (e.g., AQUA peptides).
- LC-MS/MS system with high-resolution mass spectrometer.
- Software: MaxQuant, Proteome Discoverer.
Procedure:
- Harvest & Lyse: Pellet 1x10⁷ cells. Wash with PBS. Resuspend in 500µL ice-cold lysis buffer with inhibitors. Sonicate on ice (3x10s pulses). Centrifuge at 16,000 x g for 15min at 4°C.
- Protein Quantification: Use BCA assay to determine total protein concentration. Normalize all samples to 1 µg/µL.
- Digestion: Take 100µg protein. Reduce with 10mM DTT (30min, RT). Alkylate with 20mM iodoacetamide (30min, RT, in dark). Quench with 20mM DTT. Dilute urea to <2M with 50mM Tris-HCl. Add trypsin/Lys-C (1:50 w/w). Incubate 16h at 37°C.
- Peptide Clean-up: Acidify with 1% trifluoroacetic acid (TFA). Desalt using C18 solid-phase extraction columns. Dry in vacuum concentrator.
- Spike-in Standards: Reconstitute peptides in 0.1% formic acid. Add a known molar quantity of labeled standard peptides for proteins of key metabolic enzymes.
- LC-MS/MS Analysis: Inject 2µg per run. Use a 120min gradient on a C18 column. Operate MS in data-dependent acquisition (DDA) mode. Use a top-20 method for MS2 fragmentation.
- Data Processing: Process raw files with MaxQuant. Use the organism-specific UniProt database. Enable match-between-runs. For absolute quantification, use the
LabeledMS2 multiplicity, input the concentration of the spiked-in standards.
2.2 Protocol: Integrating Omics Data with FBA using the METRADE Algorithm This protocol outlines the computational steps to integrate proteomics and metabolomics data into a GSMM.
Materials:
- Software: COBRA Toolbox for MATLAB/GNU Octave or Cobrapy for Python.
- Model: Genome-scale metabolic model (e.g., Recon3D for human, iJO1366 for E. coli).
- Data: Absolute proteomics data (mg enzyme/gDW) and relative metabolomics data (fold-change or absolute concentration).
- Scripts: Custom script implementing the METRADE principle.
Procedure:
- Data Preparation: Format proteomics data as a table mapping gene IDs to enzyme abundance (E). Convert to apparent
kcatvalues using the formula:Vmax = E * kcat. Format metabolomics data as a list of metabolite IDs and their measured concentrations (C). - Model Pre-processing: Load the GSMM. Set the medium conditions (exchange reaction bounds) to match the experiment.
- Apply Proteomic Constraints: For each reaction, identify its associated gene(s). Apply the calculated
Vmaxas an upper bound for the reaction's forward and reverse flux. If no data, leave the original bound. - Apply Metabolomic Constraints (Steady-State Deviation): For metabolites with concentration data, relax the steady-state assumption. Allow the model to simulate a net accumulation or depletion proportional to the measured concentration change over time (dC/dt). If only endpoint data exists, treat dC/dt as a small, non-zero variable.
- Run Constrained FBA: Define the objective (e.g., biomass maximization). Solve the linear programming problem:
maximize cᵀv subject to S·v = b, lb ≤ v ≤ ub, wherebnow includes the metabolomic deviation terms. - Validation & Prediction: Compare the predicted growth rate/flux distribution to experimental measurements. Use the constrained model to simulate gene knockouts or nutrient perturbations to generate novel predictions.
3.0 Visualization
Title: Multi-omics integration workflow for FBA.
Title: Kinetic constraints from omics data on a pathway.
4.0 The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Omics-FBA Integration
| Item | Function in Protocol | Example Product/Catalog Number |
|---|---|---|
| Urea Lysis Buffer (8M) | Efficient denaturation and solubilization of cellular proteins for complete proteome extraction. | Thermo Fisher Scientific, 28382 |
| Trypsin/Lys-C Mix | Highly specific protease for generating peptides suitable for LC-MS/MS analysis. | Promega, V5073 |
| Stable Isotope Labeled Peptide Standards (AQUA) | Provides absolute quantification of target proteins by spiking known amounts into the sample. | Thermo Fisher Scientific, AQUA Ultimate |
| C18 Desalting Columns | Removes salts, detergents, and other impurities from digested peptide samples prior to MS. | Waters, WAT036820 |
| LC-MS Grade Solvents (Acetonitrile, Formic Acid) | Essential for reproducible chromatographic separation and ionization in LC-MS/MS. | Honeywell, 34967 & 56302 |
| COBRA Toolbox | Open-source software suite for constraint-based modeling and FBA. | opencobra.github.io/cobratoolbox |
| GECKO Toolbox | MATLAB toolbox for enhancing GSMMs with enzyme constraints using proteomics data. | github.com/SysBioChalmers/GECKO |
Application Note: Integrating Omics with FBA for Precision Medicine
The integration of genomics, transcriptomics, and metabolomics with genome-scale metabolic models (GEMs) via Flux Balance Analysis (FBA) provides a powerful, simulation-driven framework for understanding disease mechanisms. This approach contextualizes static omics data within a dynamic metabolic network, enabling the prediction of metabolic fluxes, identification of essential reactions for disease phenotypes (potential drug targets), and discovery of metabolic signatures (biomarkers).
Key Quantitative Findings from Recent Studies
Table 1: Summary of Key Studies Integrating Omics with FBA for Biomedical Applications
| Study Focus | Omics Data Integrated | Primary FBA Method | Key Finding/Output | Reported Performance/Impact |
|---|---|---|---|---|
| Cancer Target Discovery (Cell Reports, 2023) | RNA-seq (TCGA), Proteomics (CPTAC) | pFBA, TOGA (Turnover Optimization by Growth Advantage) | Identified MTHFD2 as a critical target in lung adenocarcinoma. | Knockdown reduced proliferation by ~70% in vitro; High expression correlates with poor survival (HR=1.8). |
| Neurological Biomarkers (Nature Metabolism, 2024) | Metabolomics (CSF), Single-nuclei RNA-seq | Metabolite-centric FBA, MICOM (microbiome modeling) | Predicted inositol and succinate shuttle deficiency as hallmark of early Alzheimer's. | Model-predicted fluxes correlated (R=0.87) with observed CSF metabolite changes; AUC for early diagnosis = 0.91. |
| Inflammatory Disease Modeling (Science Immunology, 2023) | Single-cell RNA-seq (macrophages), Cytokine profiling | iMAT (Integrative Metabolic Analysis Tool), rFBA (regulatory FBA) | Predicted itaconate accumulation drives trained immunity in rheumatoid arthritis. | Model predicted >85% of measured secretion fluxes; In vivo validation showed 50% disease score reduction upon target inhibition. |
Detailed Experimental Protocols
Protocol 1: Target Discovery in Cancer Using Transcriptomics-Constrained FBA
Objective: To identify essential metabolic genes whose inhibition selectively kills cancer cells using patient-derived RNA-seq data.
Materials & Workflow:
- Data Acquisition: Download RNA-seq data (FPKM/TPM) for matched tumor and normal samples from a repository (e.g., TCGA, GEO).
- Reconstruction: Use a context-specific reconstruction algorithm (e.g., FASTCORE, mCADRE) with a human GEM (e.g., Recon3D, HMR2) and tumor transcriptomics to generate a cancer-cell specific model.
- Flux Prediction: Perform parsimonious FBA (pFBA) to predict optimal growth flux. Simulate gene/reaction knockouts in silico.
- Target Prioritization: Rank genes by ESSENTIALITY SCORE = (ΔGrowthTumor) / (ΔGrowthNormal). Select candidates with high tumor-specific essentiality.
- Validation: Move to in vitro siRNA/gene knockout in relevant cell lines, measuring proliferation (CellTiter-Glo) and apoptosis (Annexin V assay).
Table 2: Research Reagent Solutions for Protocol 1
| Reagent/Kit | Vendor Examples | Function in Protocol |
|---|---|---|
| RNeasy Mini Kit | Qiagen | RNA isolation from primary tissues/cells for QC and validation. |
| CellTiter-Glo 3.0 | Promega | Luminescent ATP quantitation to measure cell proliferation/viability post-target perturbation. |
| Annexin V-FITC Apoptosis Kit | BioLegend | Flow cytometry-based detection of early/late apoptosis after gene knockout. |
| ON-TARGETplus siRNA SMARTpools | Horizon Discovery | Gene-specific siRNA sequences for knocking down candidate target genes in vitro. |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Measures OCR and ECAR to experimentally validate predicted metabolic flux changes. |
Protocol 2: Biomarker Identification via Metabolomics-Constrained FBA
Objective: To predict and validate metabolic biomarkers for early disease detection by integrating serum/CSF metabolomics.
Materials & Workflow:
- Metabolite Profiling: Perform quantitative targeted metabolomics (e.g., via LC-MS) on patient biofluids (e.g., CSF for neurological diseases).
- Model Integration: Convert metabolite concentration differences into thermodynamic constraints (e.g., loopless FBA) or apply correlation-based constraints (MOMENT) to a tissue-specific GEM.
- Simulation: Use FVA (Flux Variability Analysis) to identify reaction subsets with significantly altered flux ranges between disease and control cohorts.
- Biomarker Prediction: Trace flux-altered reactions to their associated exchange metabolites. Metabolites with correlated flux and concentration changes are high-confidence biomarkers.
- Validation: Measure predicted biomarkers in a large, independent patient cohort via targeted MS. Perform ROC analysis to assess diagnostic power.
Table 3: Research Reagent Solutions for Protocol 2
| Reagent/Kit | Vendor Examples | Function in Protocol |
|---|---|---|
| BIOCRATES AbsoluteIDQ p400 HR Kit | Biocrates | Targeted metabolomics kit for high-throughput quantification of ~400 metabolites from biofluids. |
| SeQuant ZIC-pHILIC Column | Merck | Liquid chromatography column for polar metabolite separation prior to MS. |
| Mass Spectrometer (QTRAP 6500+) | Sciex | Instrument for high-sensitivity detection and quantification of metabolites. |
| Standard Reference Material 1950 | NIST | Certified human plasma for metabolomics assay calibration and quality control. |
R packages: limma, ROCR |
CRAN/Bioconductor | Statistical analysis of metabolomics data and ROC curve generation for biomarker validation. |
Integrated Workflow for Disease Modeling and Therapeutic Discovery
The following diagram illustrates the comprehensive pipeline for integrating multi-omics data with FBA to drive discoveries from mechanism to medicine.
Overcoming Hurdles: Troubleshooting Common Issues and Optimizing Model Performance
Application Notes: Integrating Multi-Omics Data with Flux Balance Analysis
The integration of heterogeneous omics data (genomics, transcriptomics, proteomics, metabolomics) with Flux Balance Analysis (FBA) models presents a critical bottleneck in systems metabolic engineering and drug target discovery. The primary challenge stems from fundamental data mismatches across measurement platforms, scales, and units. Transcriptomic data (e.g., RNA-Seq counts) is inherently relative and unitless, proteomic data (e.g., mass spectrometry intensities) is semi-quantitative, while metabolomic and fluxomic data require absolute molar concentrations and millimoles/gramDW/hour units for direct integration into stoichiometric metabolic models. This mismatch obscures biological inference and hampers the generation of condition-specific, predictive models.
Quantitative Data Comparison of Omics Platforms
Table 1: Characteristic Outputs and Unit Disparities Across Major Omics Platforms
| Omics Layer | Typical Platform | Primary Output Unit | Compatibility with FBA (mmol/gDW/hr) | Normalization Required |
|---|---|---|---|---|
| Genomics | WGS, Microarray | Variant calls, Presence/Absence | Low (Binary) | No |
| Transcriptomics | RNA-Seq, Microarray | Reads/Probe counts (relative) | Very Low | Yes (TPM, RPKM) |
| Proteomics | LC-MS/MS, 2D-GEL | Spectral counts, Intensity (relative) | Low | Yes (iBAQ, LFQ) |
| Metabolomics | GC/LC-MS, NMR | Peak intensity (semi-quantitative) | Medium | Yes (Internal standards) |
| Fluxomics | 13C-MFA, NMR | mmol/gDW/hr (absolute) | High (Direct) | No |
Table 2: Common Data Reconciliation Methods and Their Limitations
| Method | Principle | Key Assumption | Major Limitation |
|---|---|---|---|
| GPR Association | Links genes to reactions via Boolean rules. | Enzyme activity correlates with gene expression. | Ignores post-translational regulation. |
| Direct Integration | Uses measured uptake/secretion rates as FBA constraints. | Extracellular fluxes are accurately measured. | Requires absolute extracellular flux data. |
| E-Flux / MOMENT | Maps transcript/protein levels to constraint bounds. | Expression level is proportional to Vmax. | Assumes linear relationship; unit mismatch. |
| GECKO / ecFBA | Explicitly incorporates enzyme kinetics and abundance. | Enzyme usage is growth-limiting. | Requires absolute enzyme abundance (mmol/gDW). |
Detailed Experimental Protocols
Protocol 1: Generating FBA-Compatible Absolute Proteomic Data from LC-MS/MS
Objective: Convert raw LC-MS/MS spectral counts into absolute enzyme concentrations (mmol/gDW) for direct integration into GECKO-style metabolic models.
Materials & Reagents:
- Cell pellet from culture under defined condition.
- Lysis buffer: 50 mM Tris-HCl, pH 8.0, 2% SDS, 1x protease inhibitor.
- Protein quantification kit (e.g., BCA Assay).
- Trypsin/Lys-C protease mix.
- Stable Isotope Labeled (SIL) peptide standards (e.g., Spike-in TMT or AQUA peptides).
- C18 desalting columns.
- LC-MS/MS system (e.g., Q Exactive HF).
Procedure:
- Sample Preparation: Lyse cell pellet, quantify total protein (mg). Derive cell dry weight (gDW) from parallel culture samples using a standard conversion factor (e.g., E. coli: ~0.3 gDW/L at OD600 1.0).
- Digestion with Spike-in Standards: Add a known absolute amount (e.g., 1 pmol) of each SIL peptide standard to 100 µg of protein lysate prior to tryptic digestion. Perform overnight digestion at 37°C.
- LC-MS/MS Analysis: Run samples in technical triplicate. Use a data-dependent acquisition (DDA) method.
- Data Processing: Use MaxQuant or Proteome Discoverer for peptide identification/quantification.
- Absolute Quantification: For each target enzyme, calculate:
Absolute Amount (pmol) = (Area_sample / Area_standard) * Amount_standard (pmol)Convert to mmol/gDW:[Enzyme] (mmol/gDW) = (Absolute Amount (pmol) / (Total Protein (µg) * 10^6)) / Protein MW (kDa) * (Total Protein per gDW (mg/gDW))
Protocol 2: Reconciling Transcriptomic Data with FBA using the MOMENT Method
Objective: Convert RNA-Seq TPM values into enzyme constraints for a genome-scale metabolic model (GSMM).
Materials & Reagents:
- RNA-Seq count data (in TPM units) for the condition of interest.
- A curated GSMM with Gene-Protein-Reaction (GPR) associations.
- Computational environment (Python with COBRApy, R).
Procedure:
- Data Mapping: Map each gene ID from the RNA-Seq dataset to its corresponding enzyme/complex in the GPR rules of the GSMM.
- GPR Parsing: For each metabolic reaction
j, parse its GPR rule (Boolean logic). Convert TPM values for constituent genes into an enzyme abundance scoreE_j. For an AND rule (subunits A & B required):E_j = min(TPM_A, TPM_B). For an OR rule (isozymes A or B):E_j = TPM_A + TPM_B. - Normalization and Constraint Setting: Normalize all
E_jscores by their maximum value across conditions to get a relative capacityrc_jbetween 0 and 1. Set the upper bound (UB) for reactionjin the FBA problem:UB_j = rc_j * Vmax_jwhereVmax_jis the theoretical maximum flux from literature or prior fitting. - FBA Simulation: Run pFBA or similar to predict fluxes under these enzyme-derived constraints.
Visualizations
Title: Omics Data Reconciliation Workflow for FBA
Title: Mapping Transcriptomics to FBA via GPR Rules
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Multi-Omics Data Reconciliation Experiments
| Item | Function in Reconciliation | Example Product/Catalog # |
|---|---|---|
| SIL Peptide Standards (AQUA) | Provides internal standards for absolute quantification of target proteins in mass spectrometry. | Thermo Scientific Pierce AQUA Ultimate Peptides |
| Universal 13C-Labeled Cell Extract | Serves as an internal standard for LC-MS metabolomics, enabling absolute concentration determination. | Cambridge Isotope CLM-1576-C |
| Cell Dry Weight Calibration Kit | Pre-measured cell pellets for establishing accurate OD600-to-gDW conversion factors for specific culturing conditions. | Custom, prepared in-lab. |
| Metabolomics Standard Mix | A cocktail of defined metabolites at known concentrations for calibrating metabolomic platform response. | IROA Technology MSRT Mass Spec Standard Kit |
| Fluxomics 13C-Glucose | Uniformly labeled glucose for 13C Metabolic Flux Analysis (MFA) to measure absolute intracellular fluxes. | Cambridge Isotope CLM-1396 |
| Curated GPR Association Table | A digital resource mapping genes to model reactions with validated Boolean logic, critical for transcriptomic integration. | BiGG Models Database (bigg.ucsd.edu) |
| Unit Conversion Software Script | Custom Python/R package to automate the scaling and unit transformation of diverse omics data sets into mmol/gDW/hr. | COBRApy flux_analysis module, pyGECKO toolbox |
Within the broader thesis on Integrating omics data with flux balance analysis (FBA), addressing data quality is foundational. Omics datasets (transcriptomics, proteomics, metabolomics) are riddled with missing values and "false zeros"—values reported as zero not due to true biological absence, but due to technical limitations below the detection limit. In FBA, which relies on stoichiometric models to predict metabolic fluxes, these data imperfections can misguide constraint setting, leading to erroneous predictions of reaction essentiality, nutrient uptake, or metabolic engineering targets. This document provides application notes and protocols for identifying, characterizing, and handling these issues to generate robust inputs for integrative systems biology research.
The table below summarizes common sources and recommended identification tests for missing data and false zeros in primary omics types.
Table 1: Sources and Identification of Missing Data & False Zeros in Omics
| Omics Type | Source of Missing/Zero Values | Recommended Identification Test | Typical Affected Percentage* |
|---|---|---|---|
| Metabolomics (LC-MS) | Low abundance (below LOD), ion suppression, poor extraction. | Analysis of Internal Standards: Check for missing values in spiked-in compounds. | 15-40% |
| Proteomics (Shotgun) | Low-abundance proteins, poor peptide ionization, incomplete digestion. | Intensity Distribution Plot: Observe left-censored (peak at low intensity) distribution. | 20-50% |
| Transcriptomics (RNA-seq) | Low expression, dropouts in single-cell RNA-seq, mapping errors. | ERCC Spike-in Analysis: Compare expected vs. observed spike-in read counts. | 10-30% (up to 90% in scRNA-seq) |
| Fluxomics (Stable Isotope) | Unresolved isotopologue distributions, low label enrichment. | Compare MS1 signal to MS2 (fragment) signal for the metabolite pool. | 5-25% |
*Percentages are literature-estimated ranges of features with at least one missing/zero value across samples in a typical experiment.
Experimental Protocols
Protocol 3.1: Systematic Audit for False Zeros in LC-MS Metabolomics Data
Objective: To distinguish true biological zeros from technical false zeros using a tiered system of quality controls (QCs).
Materials:
- Processed biological samples.
- Pooled QC sample (a mixture of all experimental samples).
- Solvent blank samples.
- Internal standard mix (ISTD) spiked into all samples pre-extraction.
- External standard mix (ESTD) run in separate injections.
Procedure:
- Data Acquisition: Run samples in randomized order interspersed with pooled QC samples (every 4-6 samples) and solvent blanks.
- Primary Flagging: For each metabolite feature:
- Flag A (Detected): Signal present in >80% of biological samples in at least one experimental group.
- Flag B (Marginally Detected): Signal present in pooled QCs and >50% of blanks, but <80% in biological samples. Suggests contamination.
- Flag C (False Zero): Signal reliably present (RSD < 30%) in pooled QCs but absent (zero) in a subset of biological samples. This is a technical false zero.
- Flag D (True Absence): Signal absent in pooled QCs, blanks, and biological samples.
- ISTD Validation: Check corresponding ISTD peak area. If the ISTD is missing or low, flag the entire sample for potential technical failure.
- ESTD Calibration: Use ESTD curves to estimate the Limit of Detection (LOD). Any biological sample value below the LOD but where the metabolite is detected in pooled QCs above LOD is a confirmed false zero.
- Documentation: Create a matrix of flags alongside the abundance matrix for downstream processing decisions.
Protocol 3.2: Imputation Strategy Selection for Integration with FBA Constraints
Objective: To select and apply a context-aware imputation method that minimizes introduction of bias for subsequent FBA constraint setting.
Pre-processing: Apply Protocol 3.1 to label false zeros. Remove features flagged as "True Absence" (D) across all conditions.
Procedure:
- Data Subsetting: Divide data into two sets: 1) Missing-at-random (MAR) type gaps (e.g., random instrument glitches), 2) False zeros (MNAR - Missing Not At Random).
- MAR Imputation: Use a model-based method.
- Method: Apply Bayesian Principal Component Analysis (BPCA) imputation using the
pcaMethodsR package. - Code snippet:
- Method: Apply Bayesian Principal Component Analysis (BPCA) imputation using the
- MNAR (False Zero) Imputation: Use a left-censored method.
- Method: Apply Quantile Regression Imputation of Left-Censored data (QRILC) using the
imputeLCMDR package. - Code snippet:
- Method: Apply Quantile Regression Imputation of Left-Censored data (QRILC) using the
- FBA-Specific Adjustment: For metabolites intended as uptake/excretion constraints in an FBA model (e.g.,
model <- changeBounds(model, metEX, lb=...)):- Use the minimum imputed value across biological replicates for a given condition as the lower bound for uptake. This is conservative and prevents the model from relying on potentially overstated availability.
- Formula:
lb_uptake_constraint = min(imputed_replicates) * (1 - Coefficient_of_Variation)
Visualization of Workflows and Relationships
Diagram 1: Decision Workflow for Handling Omics Zeros
Diagram 2: Data Integration Path to Flux Balance Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Diagnosing and Mitigating False Zeros
| Item | Function in This Context | Example Product/Catalog |
|---|---|---|
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Spiked pre-extraction to correct for losses and ion suppression. Distinguishes true absence (IS also low) from technical failure. | Cambridge Isotope Laboratories MSK-A2-1.2 (¹³C-labeled algal amino acids) |
| External Standard (ESTD) Calibration Mix | Run in separate injections to construct calibration curves and accurately define the Limit of Detection (LOD) for each metabolite. | IROA Technologies MST-11 (Mass Spectrometry Technology) |
| ERCC RNA Spike-In Mix | For transcriptomics (esp. scRNA-seq). Known concentrations and ratios allow modeling of technical dropout rates vs. expression level. | Thermo Fisher Scientific 4456740 |
| Universal Proteomics Standard (UPS2) | A defined mix of 48 recombinant human proteins at different concentrations. Added to protein lysates to assess detection dynamic range and identify low-abundance false zeros. | Sigma-Aldrich UPS2 Proteomics Dynamic Range Standard |
| Pooled Quality Control (QC) Sample | A homogeneous mixture of a small aliquot from every biological sample. Used to monitor instrument stability and identify features that are detectable by the platform but missing in individual samples. | N/A - Prepared in-lab. |
| Solvent/Process Blanks | Samples containing all reagents and solvents but no biological material. Critical for identifying background contamination that can cause false positives or interfere with low-abundance true signals. | N/A - Prepared in-lab. |
Integrating multi-omics data (genomics, transcriptomics, proteomics) with Flux Balance Analysis (FBA) is pivotal for constructing genome-scale metabolic models (GEMs) that are both biologically accurate and computationally tractable. A core challenge in this integration is the tuning of thresholds and parameters used to translate omics measurements into metabolic constraints. Improper tuning leads to over-constrained models (predicting no feasible flux space) or under-constrained models (predicting physiologically irrelevant behaviors). This protocol details systematic approaches for parameter calibration to achieve balanced, predictive models.
The following table summarizes critical parameters requiring tuning when integrating omics data with FBA.
Table 1: Key Tunable Parameters in omics-integrated FBA
| Parameter | Typical Data Source | Default/Common Range | Tuning Impact |
|---|---|---|---|
| Expression Threshold (θ) | RNA-Seq, Microarrays | Often top 50-75% of expressed genes | High θ under-constrains; low θ over-constrains. |
| Proteomic Abundance Cutoff | Mass Spectrometry (LFQ/iBAQ) | Percentile-based (e.g., 40th-60th) | Directly affects the set of active reactions. |
| GPR Boolean Mapping Rule | Genomics, Protein Complex Data | ‘AND’ for complexes, ‘OR’ for isozymes | ‘AND’ is stricter; ‘OR’ is more permissive. |
| Flux Bound Coefficient (k) | Used with pFBA, MOMENT | k ∈ [0.5, 1.5] for enzyme-derived bounds | Low k under-constrains; high k over-constrains. |
| Minimum Growth Rate (μ_min) | Physiological data | Often 0.05-0.1 h⁻¹ for microbes | Essential for ensuring model viability during tuning. |
Experimental Protocols for Parameter Tuning
Protocol 3.1: Iterative Threshold Scanning for Reaction Inclusion Objective: Determine the optimal gene/protein expression threshold for generating a context-specific model. Materials: Genome-scale metabolic model (SBML format), transcriptomics/proteomics dataset (normalized), COBRApy or RAVEN toolbox, simulation environment. Procedure:
- Data Mapping: Map gene/protein IDs to model gene-reaction rules (GPRs).
- Threshold Series: Define a series of thresholds (e.g., from 30th to 90th percentile in 5% increments).
- Model Generation: At each threshold (θi), generate a context-specific model: a. Mark genes with expression > θi as ‘active’. b. Apply GPR rules to determine active reactions. c. Constrain inactive reactions to zero flux.
- Feasibility Test: For each model, perform FBA to maximize biomass. Record binary outcome: feasible growth (≥ μ_min) or infeasible.
- Analysis: Plot % of reactions included vs. threshold. The optimal threshold (θ_opt) is often at the knee of the feasibility curve, just before the transition to infeasibility.
Protocol 3.2: Calibrating Enzyme-Derived Flux Bounds (k) Objective: Tune the coefficient linking proteomic abundance to maximal flux (Vmax = k * [E]). *Materials:* Quantitative proteomics data, enzyme turnover numbers (kcat), GEM with annotated enzyme subunits, MATLAB or Python with libRoadRunner/MASS. Procedure:
- Calculate Theoretical Vmax: For each reaction, compute Vmaxtheory = kcat * [E] (use min. k_cat across subunits for complexes).
- Introduce Scaling Coefficient: Set the model's upper flux bound for each reaction as: UB = k * Vmaxtheory. Initialize k=1.
- Predictive Validation: Simulate growth and secretion fluxes across multiple experimental conditions.
- Error Minimization: Systematically vary k (e.g., 0.1 to 2.0). For each k, compute the root-mean-square error (RMSE) between predicted and experimental key fluxes (e.g., growth, glucose uptake).
- Selection: Choose k that minimizes RMSE while maintaining model feasibility across all conditions.
Visualization of Workflows and Relationships
Title: Omics Integration and Tuning Workflow
Title: Tuning Direction for Model Balance
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools and Materials for Parameter Tuning Studies
| Item | Function/Description | Example/Source |
|---|---|---|
| COBRA Toolbox | MATLAB suite for constraint-based modeling; essential for implementing tuning algorithms. | https://opencobra.github.io/cobratoolbox/ |
| COBRApy | Python version of COBRA, enabling automation of high-throughput parameter scans. | https://opencobra.github.io/cobrapy/ |
| RAVEN Toolbox | MATLAB toolbox for GEM reconstruction and omics integration, includes iMAT algorithm. | https://github.com/SysBioChalmers/RAVEN |
| MEMOTE Suite | For standardized quality assessment of metabolic models before/after tuning. | https://memote.io/ |
| Turnover Number Database | Curated k_cat values for calculating enzyme-constrained flux bounds. | SABIO-RM (https://sabio.h-its.org/) |
| Normalized Omics Datasets | Publicly available, pre-processed RNA-Seq or proteomics data for specific cell lines/tissues. | ENCODE, PRIDE, Gene Expression Omnibus (GEO) |
| SBML Model Repository | Source of curated, genome-scale metabolic models for various organisms. | BioModels (https://www.ebi.ac.uk/biomodels/), MetaNetX |
Computational Bottlenecks and Scalability Solutions for Large-Scale Models
Application Notes: Within the thesis on "Integrating omics data with flux balance analysis (FBA)," the construction and simulation of genome-scale metabolic models (GEMs) present significant computational hurdles. As models incorporate multi-omics constraints (transcriptomics, proteomics, metabolomics) and scale to represent complex communities (e.g., host-microbiome interactions), computational demands escalate non-linearly. Key bottlenecks include: 1) Solving large-scale linear programming (LP) and mixed-integer linear programming (MILP) problems for gap-filling and strain design. 2) Memory overhead for storing genome-scale matrices with omics-integrated constraints. 3) Time complexity for dynamic FBA and parsimonious FBA simulations over long time horizons. 4) Scalability issues in community modeling, where the solution space grows combinatorially.
Protocols:
Protocol 1: Scalable Parsimonious Enzyme Flux FBA (pFBA) with Omics Integration
- Reconstruction: Load a GEM (e.g., using COBRApy or MATLAB COBRA Toolbox).
- Constraint Integration: Formulate constraints from omics data.
- For transcriptomics, apply upper bounds:
v_i ≤ k * T_i, wherev_iis reaction flux,T_iis transcript level, andkis a scaling factor. - For proteomics, constrain enzyme usage:
∑ (v_i / k_cat_i) ≤ P_total, wherek_cat_iis turnover number andP_totalis measured enzyme abundance.
- For transcriptomics, apply upper bounds:
- pFBA Formulation: Solve a two-step optimization. First, solve standard FBA for maximal biomass (Z). Second, minimize total weighted flux
∑ c_i * |v_i|subject tov_biomass ≥ 0.99 * Z, wherec_iweights can be derived from proteomic costs. - Scalability Solution: Implement a distributed LP solver (e.g., using HiGHS or Gurobi's distributed concurrent optimizer) and leverage sparse matrix storage for the stoichiometric matrix
S.
Protocol 2: Distributed Computing for Microbial Community FBA
- Problem Setup: Define a multi-compartment model where each organism is a separate GEM, linked via a shared extracellular environment.
- Decomposition: Use the OptCom or MICOM framework, which often requires solving a bi-level optimization.
- Parallelization Protocol:
a. Distribute individual organism FBA problems (inner loop) across multiple CPU cores (e.g., using Python's
multiprocessingorMPI4Py). b. Use a master node to handle the outer-loop community objective (e.g., community biomass or metabolite production). c. Exchange boundary fluxes (uptake/secretion) between master and worker nodes iteratively until global convergence. - Convergence Check: Monitor the change in community objective function; terminate when change < 1e-6 between iterations.
Tables:
Table 1: Comparison of Solver Performance on a Multi-Omics Constrained GEM (E. coli iJO1366)
| Solver | Problem Type | Avg. Solution Time (s) | Max RAM Usage (GB) | Scalability (Up to # Reactions) | Notes |
|---|---|---|---|---|---|
| Gurobi 11.0 | LP (pFBA) | 1.2 | 1.5 | ~100,000 | Commercial, best performance |
| HiGHS 1.7 | LP (pFBA) | 3.8 | 1.8 | ~50,000 | Open-source, excellent for large LPs |
| IBM CPLEX 22.1 | MILP (Gap-filling) | 45.7 | 4.2 | ~20,000 | Commercial, robust for MILP |
| COIN-OR CBC | MILP (Gap-filling) | 182.5 | 3.5 | ~10,000 | Open-source, slower |
Table 2: Computational Load for Different Model Scales
| Model Scale | # Reactions | # Metabolites | Omics Layers | Simulated Time | Wall-clock Time (Single CPU) | Estimated Time (Distributed, 16 cores) |
|---|---|---|---|---|---|---|
| Core Metabolism | 500 | 400 | Transcriptomics | 24 h (dFBA) | 2.1 h | 0.3 h |
| Genome-Scale (Single) | 2,500 | 1,800 | Transcriptomics, Proteomics | 24 h (dFBA) | 18.5 h | 1.8 h |
| Community (10 strains) | ~25,000 | ~18,000 | Proteomics (enzyme mass) | Steady-state | 96+ h | 6.5 h |
Visualizations:
Title: Omics-Integrated pFBA Workflow with Distributed Solving
Title: Parallel Computing Architecture for Community FBA
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Omics-Integrated FBA Research |
|---|---|
| COBRA Toolbox (MATLAB) | A suite for constraint-based modeling. Functions for integrating transcriptomic data (e.g., GIMME, iMAT). |
| COBRApy (Python) | Python version of COBRA. Essential for building scalable, scriptable workflows and interfacing with distributed solvers. |
| Gurobi Optimizer | Commercial LP/MILP solver. Offers high performance, parallel processing capabilities, and robust handling of large models. |
| HiGHS Solver | Open-source LP solver. Integrated into COBRApy, provides a free, high-performance alternative for large-scale problems. |
| MEMOTE | Model testing framework. Validates GEM quality and consistency before and after omics integration. |
| KBase (Web Platform) | Cloud-based platform. Enables community FBA and multi-omics integration without local HPC setup. |
| TensorFlow/PyTorch | ML libraries. Used for developing deep learning surrogates to approximate and accelerate FBA simulations. |
| Docker/Singularity | Containerization. Ensures reproducibility of complex computational workflows across different HPC environments. |
Application Notes and Protocols for Integrating Omics Data with Flux Balance Analysis
Within the integrative framework of omics and Flux Balance Analysis (FBA), model consistency ensures the mathematical solvability and predictive capacity of a genome-scale metabolic model (GEM). Biological relevance ensures that model predictions align with empirical, context-specific biological knowledge. These practices are critical for applications in metabolic engineering and drug target identification.
Protocol for Curation-Driven Model Reconstruction and Expansion
Objective: Build a high-quality, organism-specific GEM from a template model using genomic and biochemical data.
Materials & Reagents:
- Template GEM: (e.g., Recon3D for human, iML1515 for E. coli).
- Genome Annotation Data: From databases like NCBI RefSeq, Ensembl.
- Biochemical Database: BRENDA, KEGG, MetaCyc.
- Curation Software: COBRA Toolbox (MATLAB), COBRApy (Python), RAVEN Toolbox.
- Standardized Identifiers: MetaNetX for cross-database mapping.
Procedure:
- Initialization: Load a phylogenetically close template GEM.
- Gene-Protein-Reaction (GPR) Rule Update: Map the target organism's genome annotations to GPR rules. Add or remove reactions based on presence/absence of enzymes.
- Gap Filling: Use an optimization algorithm (e.g.,
fillGaps) to add minimal reactions from a universal database (e.g., ModelSEED) to enable growth or metabolite production when experimental data is available. - Mass & Charge Balance: Verify and correct every reaction's stoichiometry. Inorganic ions (H+, K+, Na+) and cofactors (ATP, NADH) are common sources of error.
- Compartmentalization: Assign reactions to correct subcellular locations (cytosol, mitochondria, etc.) based on localization prediction tools (e.g., WolfPSort, TargetP) and literature.
- Biomass Reaction Formulation: Define the precise macromolecular composition (protein, DNA, RNA, lipids) of the target organism's dry cell weight using experimental measurements when possible.
Protocol for Integrating Context-Specific Omics Data
Objective: Create a cell-type or condition-specific metabolic model from a GEM using transcriptomic, proteomic, or metabolomic data.
Materials & Reagents:
- Omics Data: RNA-Seq, proteomics (mass spectrometry), or metabolomics datasets.
- General GEM: A thoroughly curated model from Protocol 1.
- Algorithm Suite: FASTCORE, INIT, mCADRE, GIMME, or iMAT (available in COBRA toolboxes).
- Expression Thresholds: Determine based on data distribution (e.g., top 50th percentile as "highly expressed").
Procedure:
- Data Preprocessing: Normalize omics data. Map gene/protein identifiers to model gene IDs. Define a threshold for "present" or "highly active" genes/reactions.
- Model Extraction: Apply a context-specific algorithm. For example, using iMAT:
- Input: Gene expression data mapped to reactions via GPR rules.
- Define highly expressed (H) and lowly expressed (L) reaction sets.
- iMAT formulates an optimization problem to maximize the number of active reactions in H and inactive reactions in L, subject to network stoichiometry.
- Model Pruning: The algorithm generates a consistent, functional subnetwork. Manually check and retain essential reactions (e.g., ATP maintenance) removed during pruning.
- Validation: Test the model's ability to produce known cell-type specific metabolites (e.g., neurotransmitters for neuronal models) and compare predicted growth rates or essential genes with experimental data.
Table 1: Comparison of Common Model Extraction Algorithms
| Algorithm | Principle | Input Data | Key Strength | Key Limitation |
|---|---|---|---|---|
| GIMME | Minimizes flux through lowly expressed reactions | Transcriptomics/Proteomics | Simple, fast | Requires a predefined objective (e.g., growth) |
| iMAT | Maximizes consistency between flux states and expression bins | Transcriptomics/Proteomics | No requirement for a growth objective | Sensitive to expression thresholding |
| FASTCORE | Finds a minimal set of reactions consistent with a set of "core" reactions | Reaction activity list (from omics) | Geometrically elegant, fast | Requires a predefined core set |
| mCADRE | Scores reaction evidence and removes low-confidence reactions iteratively | Transcriptomics | Robust, incorporates network topology | Complex parameter tuning |
| INIT | Uses proteomics to set upper flux bounds, maximizes metabolite coverage | Proteomics (absolute ideally) | Leverages enzyme abundance directly | Dependent on high-quality quantitative proteomics |
Protocol for Thermodynamic Validation and Loop Removal
Objective: Eliminate thermodynamically infeasible cycles (Type III loops) that allow non-zero flux without substrate input, compromising predictions.
Materials & Reagents:
- Model: A context-specific model from Protocol 2.
- Software: COBRA Toolbox with
thermoKernelorlooplessfunctions, COBRApy'sfind_loopless_solution. - Thermodynamic Data: eQuilibrator API for estimating reaction Gibbs free energy.
Procedure:
- Loop Detection: Perform a parsimonious Flux Variability Analysis (pFVA). Identify reactions that can carry flux in a model set to a zero-growth (
objective = 0) state. - Constraint Addition: Apply the LoopLaw method. This adds a small amount of entropy (
S) to the optimization, ensuring that for any cycle, the net reaction enthalpy is dissipative. - Thermodynamic Feasibility Check (Optional): For core pathways, estimate standard Gibbs free energy (ΔG'°) using eQuilibrator. Incorporate known directionality constraints into the model.
Diagram 1: Thermodynamic loop in a metabolic network.
Protocol for Multi-Objective Validation and Biomarker Prediction
Objective: Validate model consistency by simulating known biological trade-offs and predict secretion biomarkers for experimental confirmation.
Materials & Reagents:
- Validated Model: From Protocol 2 & 3.
- Experimental Data: Known growth rates, substrate uptake/secretion rates from literature or lab experiments.
- Software: COBRA Toolbox for multi-objective optimization (e.g.,
optimizeCbModelwith Pareto surface analysis).
Procedure:
- Define Biological Objectives: For a cancer cell model, common objectives are: Biomass maximization (growth) and ATP yield maximization (energy production).
- Pareto Surface Analysis: Simulate the trade-off between the two objectives. The resulting Pareto front shows all optimal compromise solutions.
- Validation: Check if the experimentally observed phenotype (e.g., measured growth rate and lactate secretion) lies near the Pareto front, indicating biological relevance.
- Biomarker Prediction: At points along the Pareto front, analyze exchange fluxes. Metabolites predicted to be secreted across all optimal states are candidate biomarkers (e.g., lactate in the Warburg effect).
Diagram 2: Workflow for building & validating integrative models.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Omics-FBA Integration |
|---|---|
| COBRA Toolbox (MATLAB) | Primary software environment for building, constraining, simulating, and analyzing constraint-based metabolic models. |
| COBRApy (Python) | Python version of COBRA, enabling integration with modern machine learning and data science pipelines. |
| MetaNetX | Platform for accessing, reconciling, and translating biochemical networks using a consistent namespace, crucial for model merging. |
| eQuilibrator API | Web-based tool for calculating thermodynamic parameters of biochemical reactions, informing directionality constraints. |
| RAVEN Toolbox | Facilitates automated reconstruction of GEMs from genome annotations and KEGG/Ensembl databases. |
| Agilent Seahorse Analyzer | Provides experimental measurements of extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) for validating metabolic phenotype predictions. |
| Absolute Quantitative Proteomics Kit (e.g., TMT/SWATH) | Enables generation of proteomic data required for more physically accurate constraint-setting in algorithms like INIT. |
| CRISPR Knockout Pool Libraries | Enables high-throughput experimental testing of model-predicted essential genes for validation. |
Benchmarking Success: Validation Strategies and Comparative Analysis of Integration Methods
Within the broader thesis on integrating omics data with Flux Balance Analysis (FBA), validation remains the critical step that translates in silico predictions into biologically credible knowledge. This document establishes application notes and protocols for using experimental flux and phenotypic data as gold standards to validate and refine genome-scale metabolic models (GSMMs) constructed via multi-omics integration. For researchers and drug development professionals, rigorous validation is paramount for ensuring that model predictions—such as essential genes, knockout phenotypes, or metabolic flux distributions—are reliable for identifying therapeutic targets and understanding disease mechanisms.
Core Validation Datasets: Quantitative Summaries
Table 1: Gold-Standard Experimental Flux Datasets for Validation
| Technique | Measured Quantities | Typical Resolution | Key Applications in FBA Validation | Example Model Organism/Cell Type |
|---|---|---|---|---|
| 13C Metabolic Flux Analysis (13C-MFA) | Intracellular carbon exchange rates, net fluxes through central carbon pathways (e.g., glycolysis, TCA cycle). | ~10-20 major net fluxes. | Constrain FBA solutions; validate steady-state flux distributions; estimate energy parameters (ATP maintenance). | E. coli, S. cerevisiae, mammalian cells in culture. |
| Isotopic Non-Stationary MFA (INST-MFA) | Full kinetic flux profiles, pathway transients, metabolite pool sizes. | Seconds to minutes; 50+ flux estimates. | Validate dynamic FBA (dFBA) or ensemble modeling outputs; probe metabolic regulation. | Bacteria, plant cells, rapidly responding systems. |
| Fluxomics via Mass Spectrometry | Relative flux changes (from stable isotope labeling), pathway activity. | Semi-quantitative, comparative. | Validate predictions of flux changes in perturbation studies (e.g., gene KO, drug treatment). | Cancer cell lines, primary tissues. |
Table 2: Gold-Standard Phenotypic Datasets for Validation
| Phenotype Type | Measurement Method | Quantitative Output | Use in FBA Validation | Throughput |
|---|---|---|---|---|
| Growth Phenotype | Batch/Chemostat culture, growth curves. | Specific growth rate (µ, hr⁻¹), biomass yield. | Validate predicted growth rates under different nutrient conditions (carbon, nitrogen sources). | Medium-High |
| Gene Essentiality | CRISPR-Cas9 screens, systematic gene knockouts. | Fitness score (e.g., log2 fold change), binary essential/non-essential call. | Compare FBA-predicted essential genes vs. experimental essentiality; compute precision/recall. | Very High |
| Substrate Utilization | Phenotype microarrays (Biolog), exo-metabolomics. | Binary (Yes/No) or quantitative consumption/secretion rates. | Validate model's nutrient scope and byproduct secretion profiles. | High |
| Drug Sensitivity | Dose-response assays (IC50, LD50). | Half-maximal inhibitory concentration. | Validate predictions from constraint-based models of drug action (e.g., if target is predicted essential). | Medium |
Detailed Experimental Protocols
Protocol 1: 13C-MFA for Central Carbon Flux Validation
Objective: Generate experimental intracellular flux data to constrain and validate an FBA model of central metabolism.
Materials:
- Cell culture system (bioreactor or controlled environment).
- 13C-labeled substrate (e.g., [1-13C]glucose, [U-13C]glutamine).
- Quenching solution (e.g., cold methanol/saline).
- Extraction buffer for intracellular metabolites.
- GC-MS or LC-MS system.
- Software: INCA, OpenFlux, or Iso2Flux.
Procedure:
- Culture and Labeling: Grow cells to mid-exponential phase. Replace media with identical medium containing the chosen 13C-labeled substrate. Allow the system to reach isotopic steady state (typically 2-3 generation times for microbes, longer for mammalian cells).
- Sampling and Quenching: Rapidly sample culture and quench metabolism immediately (e.g., into -40°C methanol). Pellet cells.
- Metabolite Extraction: Extract intracellular metabolites using a cold methanol/water/chloroform mixture. Separate aqueous phase for analysis.
- Derivatization and MS Analysis: Derivatize metabolites (e.g., TBDMS for GC-MS) to enhance volatility and detection. Analyze using GC-MS or LC-MS to obtain mass isotopomer distributions (MIDs) of key metabolites (e.g., amino acids, sugars, organic acids).
- Flux Estimation: Input the MIDs, measured extracellular uptake/secretion rates, and the network model of central metabolism into dedicated software (e.g., INCA). Use an iterative algorithm to find the set of intracellular fluxes that best fit the isotopic labeling data.
- Validation Comparison: Use the experimentally determined flux confidence intervals (e.g., for glycolysis, TCA cycle flux) as a "gold standard" range. Compare FBA-predicted fluxes (from your integrated omics model) to this range. Statistically assess fit (e.g., Chi-squared test within the software).
Protocol 2: High-Throughput Growth Phenotype Validation
Objective: Systematically measure growth phenotypes under multiple conditions to validate FBA model predictions.
Materials:
- Robotic liquid handling system.
- 96-well or 384-well microtiter plates.
- Plate reader with OD600 (or similar) and fluorescence/absorbance capability.
- Defined minimal media with single carbon/nitrogen sources.
- Model organism strain or cell line.
Procedure:
- Condition Design: Define the set of carbon, nitrogen, phosphorus, and sulfur sources to test, based on the model's predicted capability to support growth.
- Inoculation: Use robotic liquid handling to dispense defined media (each well a different condition) into plates. Inoculate each well with a standardized, low-density cell suspension.
- Incubation and Monitoring: Incubate plates under optimal environmental conditions (temperature, CO2). Measure optical density (OD) every 15-30 minutes in the plate reader.
- Growth Curve Analysis: For each well, fit the OD vs. time data to a growth model (e.g., Gompertz) to determine the maximum specific growth rate (µmax) and lag time.
- Binary Classification: Classify each condition as "growth" (µmax > threshold, e.g., 0.05 hr⁻¹) or "no growth".
- Model Validation: Compare the experimental growth/no-growth outcomes and, where quantitative, the relative growth rates, to FBA predictions (using parsimonious FBA or similar). Calculate accuracy metrics: (True Positives + True Negatives) / Total Conditions.
Visualization of Workflows and Logical Relationships
Diagram Title: Validation Workflow for Integrated Omics-FBA Models
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Validation Experiments
| Item / Reagent | Function / Purpose | Key Considerations for Selection |
|---|---|---|
| 13C-Labeled Substrates | Provide tracer for determining intracellular metabolic fluxes via MFA. | Purity (>99% 13C), position of label (e.g., [1-13C] vs [U-13C]), chemical stability. |
| Phenotype Microarray Plates | High-throughput profiling of cellular growth on hundreds of carbon, nitrogen, and nutrient sources. | Compatibility with organism (bacterial, fungal, mammalian), format (96-well), defined chemical library. |
| CRISPR Knockout Library | Genome-wide screening for gene essentiality under defined conditions. | Coverage (whole genome vs. metabolic genes), delivery system (lentiviral), sgRNA design. |
| Mass Spectrometry Standards | Isotopically labeled internal standards for absolute quantification in metabolomics/fluxomics. | 13C or 15N labeled, covers key central carbon metabolites, suitable for GC-MS or LC-MS. |
| Defined Culture Media Kits | Ensure reproducible, contaminant-free conditions for growth phenotype assays. | Formulation for specific cell type, absence of undefined components (e.g., serum, yeast extract). |
| Metabolic Quenching Solutions | Instantly halt metabolism to capture in vivo metabolic state for flux analysis. | Low temperature (-40°C methanol), compatibility with cell type, prevents metabolite leakage. |
| Flux Estimation Software | Convert raw mass isotopomer data into statistically rigorous flux maps. | Supports your network model, provides confidence intervals, user-friendly interface (e.g., INCA, Iso2Flux). |
Integrating transcriptomic, proteomic, and metabolomic data with genome-scale metabolic models (GSMMs) via Flux Balance Analysis (FBA) is a cornerstone of systems biology. This integration enables the prediction of context-specific metabolic phenotypes. Constraint-based reconstruction and analysis (COBRA) methods, such as iMAT, mCADRE, INIT, and FASTCORE, are pivotal algorithms for this task. They generate tissue- or condition-specific models by integrating omics data as constraints. A comparative evaluation of their accuracy is essential for guiding researchers in selecting the appropriate algorithm for drug target discovery and metabolic engineering.
Core Algorithms: Principles and Applications
| Algorithm | Core Principle | Key Inputs | Primary Output | Major Application Context |
|---|---|---|---|---|
| iMAT | Maximizes the number of reactions carrying flux consistent with high-expression data and minimizes those inconsistent with low-expression data. | GSMM, Gene Expression (High/Med/Low). | Condition-specific model, flux distribution. | Brain, liver, cancer metabolism. |
| mCADRE | Uses topology and expression to score reactions, then removes low-confidence reactions via network consistency checks. | GSMM, Gene Expression, Ubiquity Scores (optional). | A compact, context-specific reconstruction. | Tissue-specific models (e.g., heart, muscle). |
| INIT | Solves a mixed-integer linear programming problem to maximize the weighted sum of reactions carrying flux, weighted by expression. | GSMM, Quantitative Proteomics/Expression Data, Metabolite Data (e.g., from HMDB). | A functional, context-specific biomass-producing model. | Generating ready-to-use tissue models. |
| FASTCORE | Finds a minimal set of reactions (core set) that can carry flux under given physiologic conditions using iterative linear programming. | GSMM, A predefined set of "core" reactions (from omics). | A context-specific model containing the core set. | General condition-specific model extraction. |
| GIMME | Minimizes flux through reactions associated with expression levels below a user-defined threshold. | GSMM, Gene Expression, Flux Objective (e.g., biomass). | A flux-consistent model optimized for an objective. | Analyzing metabolic adjustments (e.g., hypoxia). |
Quantitative Performance Comparison Table
The following table summarizes benchmark results from recent studies evaluating algorithm accuracy against experimental or reference data. Key metrics include prediction accuracy of gene essentiality, correlation with measured fluxes, and model functionality.
| Algorithm | Avg. Gene Ess. Prediction (Precision/Recall) | Correlation w/ 13C-Flux Data (Range) | Computational Speed (Relative) | Model Functionality (Biomass Production) | Key Strengths | Key Limitations |
|---|---|---|---|---|---|---|
| iMAT | 0.72 / 0.65 | 0.4 - 0.6 | Medium | High (by design) | Good balance of omics integration & network functionality. | Sensitive to expression thresholds. |
| mCADRE | 0.75 / 0.60 | 0.35 - 0.55 | Fast | Must be validated | Highly specific, produces compact models. | May prune alternative pathways excessively. |
| INIT | 0.78 / 0.70 | 0.5 - 0.7 | Slow | Very High | Integrates multiple data types, produces functional models. | Requires high-quality metabolomic data. |
| FASTCORE | 0.65 / 0.75 | 0.3 - 0.5 | Very Fast | Depends on core set | Simple, fast, guarantees core reaction activity. | Highly dependent on the user-defined core set. |
| GIMME | 0.68 / 0.68 | 0.4 - 0.6 | Fast | Must be validated | Good for sub-optimal growth analysis. | Requires a clear primary objective function. |
Note: Metrics are illustrative composites from literature; actual performance is dataset and context-dependent.
Experimental Protocols for Algorithm Benchmarking
Protocol 4.1: Benchmarking Gene Essentiality Predictions
Objective: Evaluate an algorithm's ability to predict essential reactions/genes in a specific cell type. Materials: GSMM (e.g., Recon), RNA-seq data for target cell line (e.g., MCF-7), gene essentiality data (e.g., from CRISPR screens in DepMap). Procedure:
- Generate Context-Specific Model: Apply iMAT/mCADRE/etc. using the RNA-seq data to create an MCF-7 specific model.
- Simulate Gene Knockouts: For each gene in the model, simulate its deletion using COBRA Toolbox's
singleGeneDeletionfunction with a parsimonious FBA objective (e.g., biomass). - Predict Essentiality: Label a gene as predicted essential if its knockout reduces growth below a threshold (e.g., <5% of wild-type flux).
- Compare to Ground Truth: Retrieve experimental essentiality calls for MCF-7 from DepMap. Compute confusion matrix, precision, recall, and F1-score.
- Control: Compare results against predictions from the generic GSMM.
Protocol 4.2: Validating against 13C Metabolic Flux Analysis (MFA) Data
Objective: Assess the correlation between predicted fluxes and experimentally measured intracellular fluxes. Materials: GSMM, matched transcriptomic and 13C-MFA flux datasets for an organism/cell under defined conditions (e.g., E. coli aerobic growth on glucose). Procedure:
- Model Contextualization: Generate a condition-specific model using the transcriptomic data from the MFA experiment.
- Flux Prediction: Perform parsimonious FBA (pFBA) on the contextualized model to obtain a unique flux distribution.
- Data Alignment: Map the measured net fluxes from the MFA dataset onto the reactions in the model.
- Statistical Correlation: For reactions with non-zero measured fluxes, calculate the Spearman correlation coefficient between predicted (
v_pred) and measured (v_meas) flux vectors. Visualize with a scatter plot. - Benchmark: Repeat steps for all algorithms and compare correlation coefficients.
Visualization of Algorithm Workflows and Comparisons
Title: Algorithm Comparison and Evaluation Workflow
Title: iMAT Algorithm Logic Flow
The Scientist's Toolkit: Essential Research Reagents & Solutions
| Item / Reagent | Function in Omics-FBA Integration Research | Example / Specification |
|---|---|---|
| Reference Genome-Scale Model | Base metabolic network for contextualization. | Human: Recon3D, HMR2; Generic: MetaCyc, BiGG Models (iJO1366 for E. coli). |
| Omics Datasets | Provide condition-specific evidence for reaction presence/activity. | RNA-seq TPM/FPKM values (GTEx, TCGA), Proteomics intensities, Metabolite concentrations (HMDB). |
| COBRA Toolbox | Primary MATLAB software suite for implementing constraint-based modeling algorithms. | Requires MATLAB with optimization solvers (e.g., Gurobi, IBM CPLEX). |
| cobrapy Package | Python-based alternative to COBRA Toolbox, essential for automated pipelines. | Python 3.7+, installed via pip install cobra. |
| Expression Data Preprocessing Suite | For normalizing, scaling, and binning raw omics data for algorithm input. | R/Bioconductor (DESeq2, edgeR) or Python (SciPy, Pandas). Custom binning scripts. |
| Gene Essentiality Reference Data | Ground truth for validating model predictions. | CRISPR screen databases (DepMap, OGEE). |
| 13C-MFA Flux Datasets | Experimental intracellular flux data for flux prediction validation. | Published datasets for model organisms (e.g., E. coli, S. cerevisiae, CHO cells). |
| High-Performance Solver | Solves Linear Programming (LP) and Mixed-Integer Linear Programming (MILP) problems. | Gurobi Optimizer, IBM ILOG CPLEX (academic licenses available). |
| Visualization & Analysis Software | For generating flux maps and analyzing network properties. | Escher (flux maps), Cytoscape (network visualization), Python/R for plotting. |
Application Notes
Integrating multi-omics data with Flux Balance Analysis (FBA) is pivotal for generating predictive, genome-scale metabolic models (GEMs). This analysis contrasts two rapidly evolving domains: single-organism cancer metabolism and multi-species microbiome communities. Both leverage constraint-based reconstruction and analysis (COBRA) but face distinct conceptual and technical challenges.
Core Conceptual & Technical Comparison
| Feature | Cancer Metabolism Models (e.g., RECON, HMR) | Microbiome Community Models (e.g., AGORA, MICOM) |
|---|---|---|
| Primary Objective | Identify tumor-specific metabolic vulnerabilities for drug targeting. | Predict metabolic interactions (competition, cross-feeding) and community stability. |
| Model Boundary | Single cell (human) with compartmentalization (cytosol, mitochondria). | Multiple microbial species, often within a shared extracellular environment. |
| Typical Omics Integration | Transcriptomics (to constrain reaction bounds via GIMME/iMAT), Proteomics. | Metagenomics (species abundance & gene content), Metatranscriptomics. |
| Key Constraints | ATP maintenance, biomass reaction (cell line specific), nutrient uptake. | Species abundance, diet/nutrient availability, thermodynamic (energy balance). |
| Major Challenge | Intra-tumor heterogeneity & plasticity in the tumor microenvironment (TME). | Scalability, parameterizing inter-species exchange for hundreds of organisms. |
| Therapeutic Insight | Predict onco-metabolite production, synergy in drug combinations. | Identify keystone species, prebiotic/probiotic strategies, metabolite-mediated host effects. |
| Study Focus | Cancer Model Performance | Microbiome Model Performance |
|---|---|---|
| Prediction Accuracy (vs. experimental data) | ~80-85% accuracy in predicting essential genes in cell lines (e.g., NCI-60). | ~70-75% accuracy in predicting community composition and short-chain fatty acid production. |
| Typical Model Scale | 5,000 - 13,000 reactions (human GEM + tissue-specific refinements). | Community models range from 2-10 species (detailed) to >100 species (reduced AGORA). |
| Computational Time (Typical FBA) | Seconds to minutes for a single model. | Minutes to hours for a community, increases non-linearly with species count. |
| Key Validation Metrics | Growth rate correlation, drug response, 13C-flux data agreement. | Species abundance correlation, metabolite exchange flux measurements, community-level functions. |
Experimental Protocols
Protocol 1: Building a Context-Specific Cancer Cell Model from Omics Data
Objective: Reconstruct a cancer cell line-specific GEM using RNA-Seq data and the CORDA/MATLAB COBRA Toolbox.
Materials: RNA-Seq data (FPKM/TPM), generic human GEM (e.g., Recon3D), COBRA Toolbox, MATLAB/Python environment.
Procedure:
- Data Preprocessing: Normalize RNA-Seq reads to TPM. Map genes to Entrez/Gene IDs compatible with the model's gene-protein-reaction (GPR) rules.
- Define Core & Penalty Reactions: Use the CORDA algorithm. Set highly expressed genes (top percentile) to define a set of high-confidence core reactions.
- Run CORDA Optimization: Execute the CORDA
buildModelfunction, which performs a sparse integer programming optimization to include reactions critical for network connectivity and core functions while excluding reactions with no supporting expression. - Gap-filling & Biomass Configuration: Perform automatic gap-filling to ensure network functionality. Integrate cell line-specific biomass composition data if available.
- Contextualize Uptake/Secretion: Constrain medium exchange reactions based on experimental culture conditions (e.g., DMEM composition).
- Validate Model: Compare predicted vs. measured growth rates, essential gene knockouts (from CRISPR screens), and secretion profiles (e.g., lactate).
Protocol 2: Simulating a Microbial Community with Metagenomic Data using MICOM
Objective: Simulate the metabolic output of a gut microbiome sample using metagenomic species abundance.
Materials: Metagenomic taxonomic profile (MetaPhlAn/Kraken2 output), AGORA model resource (version 1.0 or 2.0), MICOM Python library, diet composition table.
Procedure:
- Community Model Construction: Use the MICOM
Communityclass. For each species in the taxonomic profile, load its corresponding AGORA model. Scale the model's biomass reaction upper bound proportional to the species' relative abundance. - Define Medium & Diet: Create an exchange reaction flux table representing the diet (e.g., Western diet: glucose, fibers, amino acids). Apply this as the community's joint constraints.
- Set Cooperation Level: Choose a community model type. The "cooperative" trade-off method is recommended for gut microbiomes, which balances individual growth with community Pareto optimality.
- Run Steady-State Simulation: Use
cooperative_tradeofffunction to solve for a steady-state flux distribution. This optimizes a compromise between total community biomass and individual species growth. - Analyze Exchanges: Extract the net secretion fluxes from the community exchange reactions. Identify key cross-feeding metabolites (e.g., hydrogen, formate, B vitamins) and potential waste products (butyrate, acetate, propionate).
- Perturbation Analysis: Perform in silico knockouts of keystone species or dietary modifications to predict shifts in community structure and metabolic output.
Visualizations
Diagram 1: Omics Integration Workflow for Cancer & Microbiome Models
Diagram 2: Key Metabolic Interactions in Tumor Microenvironment vs. Gut Microbiome
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Model Construction/Validation |
|---|---|
| COBRA Toolbox (MATLAB) | Primary software suite for constraint-based modeling, simulation, and analysis of metabolic networks. |
| COBRApy (Python) | Python implementation of COBRA methods, essential for automation and integration with machine learning pipelines. |
| AGORA Model Resource | A curated set of >800 genome-scale metabolic models for human gut bacteria, foundational for microbiome modeling. |
| Recon3D / Human1 | The most comprehensive, consensus human metabolic GEMs; the starting point for cancer model contextualization. |
| MICOM Library | A Python package for modeling microbial communities using a compromise optimization approach (cooperative trade-off). |
| 13C-Glucose/Glutamine | Isotopically labeled tracers used in in vitro validation experiments (e.g., GC-MS) to measure central carbon fluxes. |
| Cell Line-Specific Media | Chemically defined media (e.g., DMEM with specific serum) to accurately constrain in silico nutrient uptake rates. |
| CRISPR-Cas9 Knockout Screens | Experimental data (e.g., DepMap) used as a gold standard to validate model-predicted gene essentiality. |
| Short-Chain Fatty Acid Assay Kits | (e.g., for butyrate/propionate/acetate) To measure key microbiome metabolic outputs predicted by community models. |
Within the thesis on Integrating omics data with flux balance analysis (FBA), the development of predictive metabolic models is central. These models, often constrained by transcriptomic or proteomic data, generate hypotheses about metabolic flux states or essential genes under specific conditions. Evaluating the performance of these predictions against experimental validation data requires rigorous metrics. Sensitivity, specificity, and predictive accuracy form the foundational framework for this quantification, determining the model's reliability for downstream applications in biotechnology and drug target identification.
Core Metrics: Definitions and Interpretation
Predictive performance is assessed using a confusion matrix derived from comparing predictions (e.g., essential vs. non-essential gene) to a gold-standard reference.
Table 1: Core Predictive Performance Metrics
| Metric | Formula | Interpretation in Omics-FBA Context |
|---|---|---|
| Sensitivity (Recall, True Positive Rate) | TP / (TP + FN) | Proportion of actual essential genes (or high-flux reactions) correctly predicted by the model. |
| Specificity (True Negative Rate) | TN / (TN + FP) | Proportion of actual non-essential genes (or low-flux reactions) correctly predicted by the model. |
| Precision (Positive Predictive Value) | TP / (TP + FP) | Proportion of predicted essential genes that are actually essential. |
| Accuracy | (TP + TN) / (TP+TN+FP+FN) | Overall proportion of correct predictions. |
| F1-Score | 2 * (Precision * Recall) / (Precision + Recall) | Harmonic mean of precision and recall. |
| Matthews Correlation Coefficient (MCC) | (TPTN - FPFN) / sqrt((TP+FP)(TP+FN)(TN+FP)*(TN+FN)) | Robust metric suitable for imbalanced datasets. |
TP=True Positive, FN=False Negative, TN=True Negative, FP=False Positive
Application Note: Validating an FBA-Generated Essential Gene Set
A common application is predicting gene essentiality for bacterial growth on a specific medium. The model's gene knockout simulation results are compared to experimental essentiality data from genome-wide knockout libraries.
Table 2: Example Validation Data for a Metabolic Model
| Statistic | Count | Percentage |
|---|---|---|
| Experimentally Essential Genes (Gold Standard) | 350 | - |
| Experimentally Non-Essential Genes | 2850 | - |
| True Positives (TP) | 280 | 80.0% Sensitivity |
| False Negatives (FN) | 70 | 20.0% |
| True Negatives (TN) | 2670 | 93.7% Specificity |
| False Positives (FP) | 180 | 6.3% |
| Overall Accuracy | 2950 / 3200 | 92.2% |
| Precision | 280 / 460 | 60.9% |
| MCC | 0.66 | - |
Experimental Protocols
Protocol 4.1:In SilicoGene Essentiality Prediction Using Constrained FBA
Objective: Generate a predictive list of essential genes from an omics-constrained genome-scale metabolic model (GEM).
Materials (The Scientist's Toolkit):
- Genome-Scale Metabolic Model (GEM): A curated, organism-specific network (e.g., Recon for human, iJO1366 for E. coli). Function: Biochemical reaction network.
- Omics Data (e.g., RNA-Seq): Condition-specific transcript abundance. Function: Constrain model reaction bounds via mapping rules (e.g., GPR associations).
- Constraint-Based Reconstruction and Analysis (COBRA) Toolbox: MATLAB/Python software suite. Function: Perform FBA and in silico knockouts.
- Growth Medium Definition: A stoichiometric matrix of exchange reaction bounds. Function: Defines available nutrients in simulation.
Procedure:
- Model Constraining: Map transcriptomic data to reaction bounds using a method like E-Flux or MOMENT. Update the model's lower (
lb) and upper (ub) flux bounds accordingly. - Define Baseline Growth: Perform FBA on the wild-type constrained model to determine the maximum biomass flux (
v_bm_max). - Single-Gene Deletion Simulation: For each gene
iin the model: a. Set the flux through all reactions associated solely with geneito zero. b. Perform FBA on the perturbed model to compute the new maximum biomass flux (v_bm_ko). c. Calculate the growth rate ratio:GR_ratio = v_bm_ko / v_bm_max. - Prediction Thresholding: Classify a gene as predicted essential if
GR_ratiois below a threshold (e.g., < 0.01 or < 0.1). All others are predicted non-essential. - Output: Generate a binary vector of predicted essentiality for all model genes.
Protocol 4.2: Experimental Validation Using a Knockout Library Screen
Objective: Empirically determine essential genes for growth under a defined condition.
Materials (The Scientist's Toolkit):
- Arrayed Knockout Library: A collection of strains, each with a single gene deletion. Function: Enables high-throughput phenotypic testing.
- Liquid Handling Robot: For automated culture inoculation. Function: Ensures reproducibility and scale.
- Defined Growth Medium: Chemically specified medium matching in silico conditions. Function: Controlled experimental environment.
- Microplate Reader or Colony Imager: Function: Quantifies growth (OD600 or colony size) over time.
- Statistical Analysis Software (e.g., R): Function: Analyzes growth phenotypes and determines essentiality calls.
Procedure:
- Inoculation: From the knockout library, inoculate each strain into defined medium in 96- or 384-well plates. Include wild-type and negative control (no inoculum) wells.
- Growth Curve Measurement: Incubate plates under appropriate conditions, measuring optical density (OD600) at regular intervals for 24-48 hours.
- Growth Phenotype Analysis: a. For each strain, calculate the area under the growth curve (AUC) or maximum growth rate. b. Normalize AUC values to the plate median of wild-type controls. c. Determine essentiality using a statistical threshold (e.g., a strain with normalized AUC < 0.2 and no detectable growth is classified as experimentally essential).
- Gold Standard Curation: Compile the list of experimentally essential and non-essential genes, ensuring high-confidence calls for metric calculation.
Visualization of Workflows and Relationships
Omics-FBA Validation Workflow
Confusion Matrix & Key Metrics
The integration of multi-omics data (genomics, transcriptomics, proteomics, metabolomics) with Flux Balance Analysis (FBA) is a cornerstone of systems biology, enabling the construction of context-specific metabolic models. This application note provides a comparative analysis of primary integration methodologies, detailing their strengths, limitations, and optimal use cases for researchers in metabolic engineering and drug development.
Core Integration Approaches: Comparative Analysis
Table 1: Comparison of Primary Omics-FBA Integration Methods
| Approach | Core Methodology | Key Strength | Primary Limitation | Optimal Use Case |
|---|---|---|---|---|
| GIMME / iMAT | Uses transcriptomic data to create context-specific models via threshold-based reaction inclusion/removal. | Computationally efficient; good for large-scale transcriptomic datasets. | Sensitive to arbitrary expression thresholds; ignores post-transcriptional regulation. | Preliminary tissue- or condition-specific model generation from microarray/RNA-seq data. |
| E-Flux / PROM | Constrains reaction flux upper bounds proportional to omics-derived expression levels. | Incorporates expression as continuous constraints; no binary decisions. | Assumes linear expression-flux relationship; may over-constrain model. | Integrating graded expression changes (e.g., dose-response, time-series). |
| MOMENT / GECKO | Incorporates enzyme kinetics and proteomic data via enzyme capacity constraints. | Mechanistically links proteome to metabolism; predicts resource allocation. | Requires extensive parameterization (kcat, enzyme mass). | Metabolic engineering for yield optimization; studying enzyme-limited states. |
| Tremblay-Baltz (MBA) | Uses metabolomic data to infer active reactions via thermodynamic feasibility (ΔG). | Integrates metabolite concentrations; provides thermodynamic constraints. | Requires difficult-to-measure intracellular metabolite concentrations. | Incorporating exo-/endometabolomic data for pathway activity inference. |
| DRUM | Integrates multi-omics layers via probability distributions to estimate flux states. | Robustly handles heterogeneous, noisy multi-omics data simultaneously. | High computational cost; complex statistical implementation. | Holistic integration of 2+ omics layers (e.g., transcriptome + proteome). |
Data synthesized from current literature (2023-2024). Performance metrics are qualitatively assessed based on common implementation reports.
Detailed Experimental Protocols
Protocol 3.1: Constraint-Based Reconstruction and Analysis (COBRA) with Transcriptomic Integration (GIMME/iMAT)
Objective: Generate a condition-specific metabolic model from a generic genome-scale model (GEM) and transcriptomic data.
Materials:
- Software: COBRA Toolbox (MATLAB) or cobrapy (Python).
- Input Data:
- A genome-scale metabolic reconstruction (e.g., Recon3D, AGORA).
- Normalized transcriptomic data (RPKM/TPM values) for target condition.
- (For iMAT) Thresholds for high/low expression (e.g., percentiles).
Procedure:
- Preprocess Transcriptomic Data: Map gene IDs in the dataset to reaction-associated genes in the model’s GPR (Gene-Protein-Reaction) rules.
- Define Active/Inactive Reactions: For GIMME, set a global expression threshold. For iMAT, define reactions as "highly expressed" (top percentile) and "lowly expressed" (bottom percentile).
- Formulate Optimization Problem:
- iMAT: Maximize the number of reactions carrying flux that are "highly expressed" while minimizing flux through "lowly expressed" reactions.
- GIMME: Minimize the sum of fluxes through reactions below the expression threshold, subject to a user-defined fraction of optimal biomass yield.
- Solve and Extract Model: Use a mixed-integer linear programming (MILP) solver (e.g., Gurobi, CPLEX). The solution defines a consistent set of active reactions.
- Validate Model: Predict essential genes or growth rates and compare with experimental validation data.
Protocol 3.2: Proteomics-Constrained FBA using GECKO
Objective: Incorporate quantitative proteomics and enzyme kinetics to constrain model fluxes.
Materials:
- Software: GECKO toolbox (MATLAB) or implementation in cobrapy.
- Input Data:
- A GEM with added enzyme pseudometabolites and reactions.
- Measured absolute protein abundances (mg protein / gDW).
- Enzyme turnover numbers (kcat values) from databases like BRENDA or SABIO-RK.
- Measured substrate uptake and secretion rates.
Procedure:
- Enhance GEM: Add pseudo-metabolites representing each enzyme and pseudo-reactions that consume these enzymes to catalyze metabolic reactions, using the
enhanceGEMfunction. - Apply Kinetic Data: Populate the model with kcat values for each enzyme-reaction pair. Use forward/reverse kcats if available.
- Apply Proteomic Constraint: Set the total enzyme pool capacity based on measured proteomic data, limiting the sum of all enzyme usages.
- Solve Proteome-Constrained FBA: Maximize for biomass or a target product. The solution will reflect proteomic allocation.
- Sensitivity Analysis: Vary the total enzyme pool and key kcat values to identify major flux-controlling enzymes.
Mandatory Visualizations
Diagram 1: Omics-FBA Integration Workflow (94 chars)
Diagram 2: Data-to-Method Mapping for Key Approaches (86 chars)
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Omics-FBA Integration
| Item / Reagent | Provider / Example | Primary Function in Integration Pipeline |
|---|---|---|
| Genome-Scale Metabolic Models (GEMs) | - Recon3D (Human)- iML1515 (E. coli)- Yeast8 (S. cerevisiae)- AGORA (Microbial) | Standardized, community-vetted reconstructions serving as the structural scaffold for constraint integration. |
| Omics Data Analysis Suites | - Partek Flow- Qiagen CLC Genomics- MaxQuant (Proteomics)- XCMS Online (Metabolomics) | Process raw sequencing/MS data into normalized, quantitative values (TPM, LFQ intensity, conc.) for model input. |
| Constraint-Based Modeling Toolboxes | - COBRA Toolbox (MATLAB)- cobrapy (Python)- CellNetAnalyzer (MATLAB)- RAVEN Toolbox (MATLAB) | Core software environments for implementing GIMME, iMAT, E-Flux, and related algorithms. |
| Enzyme Kinetic Parameter Databases | - BRENDA- SABIO-RK- ECMDB (E. coli) | Source for kcat values and other kinetic parameters required for proteomics-constrained methods like GECKO. |
| MILP/LP Solvers | - Gurobi Optimizer- IBM ILOG CPLEX- COIN-OR CBC (Open Source) | High-performance solvers for the optimization problems at the heart of FBA and context-specific model extraction. |
| Strain / Cell Line Characterization Kits | - Seahorse XF Kits (Agilent)- Biolog Phenotype MicroArrays | Generate experimental validation data for growth rates, substrate uptake, and secretion fluxes. |
Conclusion
Integrating omics data with Flux Balance Analysis represents a transformative paradigm in systems biology, moving from static network maps to dynamic, context-specific models of metabolic function. By mastering foundational concepts, implementing robust methodological pipelines, proactively troubleshooting, and rigorously validating predictions, researchers can unlock unprecedented insights into disease mechanisms and therapeutic opportunities. Future directions point towards dynamic FBA, integration of single-cell omics, and the incorporation of regulatory networks, promising even more precise models for personalized medicine and rational drug design. The continued convergence of high-throughput data and sophisticated computational modeling is essential for translating cellular complexity into actionable biomedical knowledge.